General Information About Childhood Soft Tissue Sarcoma
Childhood soft tissue sarcoma is a disease in which malignant (cancer) cells form in soft tissues of the body.
Soft tissues of the body connect, support, and surround other body parts and organs. The soft tissues include the following:
- Fat.
- A mix of bone and cartilage.
- Fibrous tissue.
- Muscles.
- Nerves.
- Tendons (bands of tissue that connect muscles to bones).
- Synovial tissues (tissues around joints).
- Blood vessels.
- Lymph vessels.
Soft tissue sarcoma may be found anywhere in the body. In children, the tumors form most often in the arms, legs, or trunk (chest and abdomen).
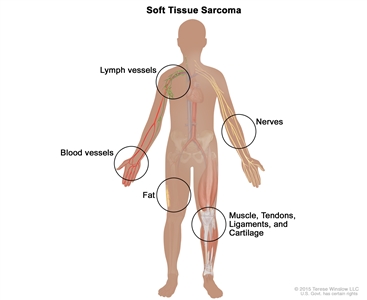
Soft tissue sarcoma forms in soft tissues of the body, including muscle, tendons, fat, blood vessels, lymph vessels, nerves, and tissue around joints.
Soft tissue sarcoma occurs in children and adults.
Soft tissue sarcoma in children may respond differently to treatment, and may have a better prognosis than soft tissue sarcoma in adults. (See the PDQ summary on Adult Soft Tissue Sarcoma Treatment for information on treatment in adults.)
Having certain diseases and inherited disorders can increase the risk of childhood soft tissue sarcoma.
Anything that increases your risk of getting a disease is called a risk factor. Having a risk factor does not mean that you will get cancer; not having risk factors doesn't mean that you will not get cancer. Talk with your child's doctor if you think your child may be at risk.
Risk factors for childhood soft tissue sarcoma include having the following inherited disorders:
- Li-Fraumeni syndrome.
- Familial adenomatous polyposis (FAP).
- Retinoblastoma 1gene changes.
- Neurofibromatosis type 1 (NF1).
- Werner syndrome.
Other risk factors include the following:
- Past treatment with radiation therapy.
- Having AIDS (acquired immune deficiency syndrome) and Epstein-Barr virus infection at the same time.
The most common sign of childhood soft tissue sarcoma is a painless lump or swelling in soft tissues of the body.
A sarcoma may appear as a painless lump under the skin, often on an arm, a leg, or the trunk. There may be no other signs or symptoms at first. As the sarcoma gets bigger and presses on nearby organs, nerves, muscles, or blood vessels, it may cause signs or symptoms, such as pain or weakness.
Other conditions may cause the same signs and symptoms. Check with your child's doctor if your child has any of these problems.
Diagnostic tests are used to detect (find) and diagnose childhood soft tissue sarcoma.
The following tests and procedures may be used:
- Physical exam and history: An exam of the body to check general signs of health, including checking for signs of disease, such as lumps or anything else that seems unusual. A history of the patient's health habits and past illnesses and treatments will also be taken.
- X-rays: An x-ray is a type of energy beam that can go through the body onto film, making pictures of areas inside the body.
- MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas of the body, such as the chest, abdomen, arms, or legs. This procedure is also called nuclear magnetic resonance imaging (NMRI).
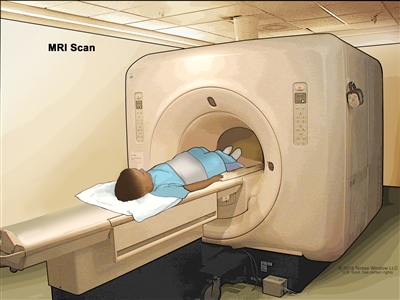
Magnetic resonance imaging (MRI) of the abdomen. The child lies on a table that slides into the MRI scanner, which takes pictures of the inside of the body. The pad on the child's abdomen helps make the pictures clearer. - CT scan (CAT scan): A procedure that makes a series of detailed pictures of areas inside the body, such as the chest or abdomen, taken from different angles. The pictures are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography.
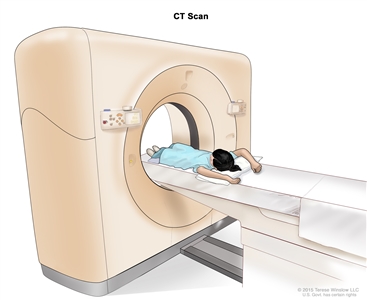
Computed tomography (CT) scan of the abdomen. The child lies on a table that slides through the CT scanner, which takes x-ray pictures of the inside of the abdomen. - Ultrasound exam: A procedure in which high-energy sound waves (ultrasound) are bounced off internal tissues or organs and make echoes. The echoes form a picture of body tissues called a sonogram. The picture can be printed to be looked at later.
If tests show there may be a soft tissue sarcoma, a biopsy is done.
One of the following types of biopsies is usually used:
The placement of needles or incisions for the biopsy can affect the success of later surgery to remove the tumor. If possible, the surgeon who will remove any tumor that is found should be involved in planning the biopsy.
In order to plan the best treatment, the sample of tissue removed during the biopsy must be large enough to find out the type of soft tissue sarcoma and do other laboratory tests. Tissue samples will be taken from the primary tumor, lymph nodes, and other areas that may have cancer cells. A pathologist views the tissue under a microscope to look for cancer cells and to find out the type and grade of the tumor. The grade of a tumor depends on how abnormal the cancer cells look under a microscope and how quickly the cells are dividing. High-grade and mid-grade tumors usually grow and spread more quickly than low-grade tumors.
Because soft tissue sarcoma can be hard to diagnose, the tissue sample should be checked by a pathologist who has experience in diagnosing soft tissue sarcoma.
One or more of the following laboratory tests may be done to study the tissue samples:
- Molecular test: A laboratory test to check for certain genes, proteins, or other molecules in a sample of tissue, blood, or other body fluid. A molecular test may be done with other procedures, such as biopsies, to help diagnose some types of cancer. Molecular tests check for certain gene or chromosome changes that occur in some soft tissue sarcomas.
- Reverse transcription-polymerase chain reaction (RT-PCR) test: A laboratory test in which cells in a sample of tissue are studied using chemicals to look for changes in the expression of certain genes. When genes are expressed they make specific proteins that are needed for the structure, function, and monitoring of the body's tissues and organs. This test is done in order to identify the type of tumor.
- Cytogenetic analysis: A laboratory test in which cells in a sample of bone marrow, blood, amniotic fluid, tumor or other tissue is viewed under a microscope to look for changes in the chromosomes. Fluorescence in situ hybridization (FISH) is a type of cytogenetic analysis.
- Immunocytochemistry: A test that uses antibodies to check for certain antigens (markers) in a sample of cells. The antibody is usually linked to an enzyme or fluorescent dye that causes the cells that have that marker to become visible under a microscope. This type of test may be used to tell the difference between different types of soft tissue sarcoma.
There are many different types of soft tissue sarcomas.
The cells of each type of sarcoma look different under a microscope. The soft tissue tumors are grouped based on the type of soft tissue cell where they first formed.
This summary is about the following types of soft tissue sarcoma:
Fat tissue tumors
- Liposarcoma. This is a rare cancer of the fat cells. Liposarcoma usually forms in the fat layer just under the skin. In children and adolescents, liposarcoma is often low grade (likely to grow and spread slowly).
There are several different types of liposarcoma. Myxoid liposarcoma is usually low grade and responds well to treatment. The cells of myxoid liposarcoma have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose myxoid liposarcoma, the tumor cells are checked for this genetic change. Pleomorphic liposarcoma is usually high grade (likely to grow and spread quickly) and is less likely to respond well to treatment.
Bone and cartilage tumors
Bone and cartilage tumors are a mix of bone cells and cartilage cells. Bone and cartilage tumors include the following types:
- Extraskeletal mesenchymal chondrosarcoma. This type of bone and cartilage tumor often affects young adults and occurs in the head and neck.
- Extraskeletal osteosarcoma. This type of bone and cartilage tumor is very rare in children and adolescents. It is likely to come back after treatment and may spread to the lungs.
Fibrous (connective) tissue tumors
Fibrous (connective) tissue tumors include the following types:
- Desmoid-type fibromatosis (also called desmoid tumor or aggressive fibromatosis). This fibrous tissue tumor is low grade (likely to grow slowly). It may come back in nearby tissues but usually does not spread to distant parts of the body. Rarely, the tumor may disappear without treatment.
Desmoid tumors sometimes occur in children with changes in the adenomatous polyposis coli (APC) gene. Changes in this gene cause familial adenomatous polyposis (FAP). FAP is an inherited condition in which many polyps (growths on mucous membranes) form on the inside walls of the colon and rectum. Genetic counseling (a discussion with a trained professional about inherited diseases and a possible need for gene testing) may be needed.
- Dermatofibrosarcoma protuberans. This is a rare tumor of the deep layers of the skin found in children and adults. The cells of this tumor have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose dermatofibrosarcoma protuberans, the tumor cells are checked for this genetic change.
- Fibrosarcoma.
There are two types of fibrosarcoma in children and adolescents:
- Infantile fibrosarcoma (also called congenital fibrosarcoma). This type of fibrosarcoma is found in children aged 4 years and younger. It most often occurs in infants and may be seen in a prenatal ultrasound exam. This tumor is often large and fast growing, but rarely spreads to distant parts of the body. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose infantile fibrosarcoma, the tumor cells are checked for this genetic change.
- Adult-type fibrosarcoma. This is the same type of fibrosarcoma found in adults. The cells of this tumor do not have the genetic change found in infantile fibrosarcoma. See the PDQ summary on Adult Soft Tissue Sarcoma Treatment for more information.
- Inflammatory myofibroblastic tumor. This is a fibrous tissue tumor that occurs in children and adolescents. It is likely to come back after treatment but rarely spreads to distant parts of the body. A certain genetic change has been found in about half of these tumors.
- Low-grade fibromyxoid sarcoma. This is a slow-growing tumor that affects young and middle-aged adults. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose low-grade fibromyxoid sarcoma, the tumor cells are checked for this genetic change. The tumor may come back many years after treatment and spread to the lungs and the lining of the wall of the chest cavity. Lifelong follow-up is needed.
- Myxofibrosarcoma. This is a rare fibrous tissue tumor that is found less often in children than in adults.
- Sclerosing epithelioid fibrosarcoma. This is a rare fibrous tissue tumor that can come back and spread to other places years after treatment. Long-term follow-up is needed.
Skeletal muscle tumors
Skeletal muscle is attached to bones and helps the body move.
- Rhabdomyosarcoma. Rhabdomyosarcoma is the most common childhood soft tissue sarcoma in children 14 years and younger. See the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.
Smooth muscle tumors
Smooth muscle lines the inside of blood vessels and hollow internal organs such as the stomach, intestines, bladder, and uterus.
- Leiomyosarcoma. This smooth muscle tumor has been linked with Epstein-Barr virus in children who also have HIV disease or AIDS. Leiomyosarcoma may also form as a second cancer in survivors of inherited retinoblastoma, sometimes many years after the initial treatment for retinoblastoma.
So-called fibrohistiocytic tumors
- Plexiform fibrohistiocytic tumor. This is a rare tumor that usually affects children and young adults. The tumor usually starts as a painless growth on or just under the skin on the arm, hand, or wrist. It may rarely spread to nearby lymph nodes or to the lungs.
Peripheral nervous system tumors
Peripheral nervous system tumors include the following types:
- Ectomesenchymoma. This is a rare, fast-growing tumor of the nerve sheath (protective covering of nerves that are not part of the brain or spinal cord) that occurs mainly in children. Ectomesenchymomas may form in the head and neck, abdomen, perineum, scrotum, arms, or legs.
- Malignant peripheral nerve sheath tumor. This is a tumor that forms in the nerve sheath. Some children who have a malignant peripheral nerve sheath tumor have a rare genetic condition called neurofibromatosis type 1 (NF1). This tumor may be low grade or high grade.
- Malignant triton tumor. These are very rare, fast-growing tumors that occur most often in children with NF1.
Pericytic (Perivascular) Tumors
Pericytic tumors form in cells that wrap around blood vessels. Pericytic tumors include the following types:
- Myopericytoma. Infantile hemangiopericytoma is a type of myopericytoma. Children younger than 1 year at the time of diagnosis may have a better prognosis. In patients older than 1 year, infantile hemangiopericytoma is more likely to spread to other parts of the body, including the lymph nodes and lungs.
- Infantile myofibromatosis. Infantile myofibromatosis is another type of myopericytoma. It is a fibrous tumor that often forms in the first 2 years of life. There may be one nodule under the skin, usually in the head and neck area (myofibroma), or nodules in several skin areas, muscle, and bone (myofibromatosis). These tumors may go away without treatment.
Tumors of unknown origin
Tumors of unknown origin (the place where the tumor first formed is not known) include the following types:
- Alveolar soft part sarcoma. This is a rare tumor of the soft supporting tissue that connects and surrounds the organs and other tissues. It is most commonly found in the limbs but can occur in the tissues of the mouth, jaws, and face. It may grow slowly and may have spread to other parts of the body at the time of diagnosis. Alveolar soft part sarcoma may have a better prognosis when the tumor is 5 centimeters or smaller or when the tumor is completely removed by surgery. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose alveolar soft part sarcoma, the tumor cells are checked for this genetic change.
- Clear cell sarcoma of soft tissue. This is a slow-growing soft tissue tumor that begins in a tendon (tough, fibrous, cord-like tissue that connects muscle to bone or to another part of the body). Clear cell sarcoma most commonly occurs in deep tissue of the foot, heel, and ankle. It may spread to nearby lymph nodes. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose clear cell sarcoma of soft tissue, the tumor cells are checked for this genetic change.
- Desmoplastic small round cell tumor. This tumor most often forms in the abdomen, pelvis or tissues around the testes, but it may form in the kidney. Desmoplastic small round cell tumor may also spread to the lungs and other parts of the body. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose desmoplastic small round cell tumor, the tumor cells are checked for this genetic change.
- Epithelioid sarcoma. This is a rare sarcoma that usually starts deep in soft tissue as a slow growing, firm lump and may spread to the lymph nodes.
- Extrarenal (extracranial) rhabdoid tumor. This is a rare, fast-growing tumor of soft tissues such as the liver and peritoneum. It usually occurs in young children, including newborns, but it can occur in older children and adults. Rhabdoid tumors may be linked to a change in a tumor suppressor gene called SMARCB1. This type of gene makes a protein that helps control cell growth. Changes in the SMARCB1 gene may be inherited (passed on from parents to offspring). Genetic counseling (a discussion with a trained professional about inherited diseases and a possible need for gene testing) may be needed.
- Extraskeletal myxoid chondrosarcoma. This is a rare soft tissue sarcoma that may be found in children and adolescents. Over time, it tends to spread to other parts of the body, including the lymph nodes and lungs. The cells of this tumor usually have a genetic change, often a translocation (part of one chromosome switches places with part of another chromosome). In order to diagnose extraskeletal myxoid chondrosarcoma, the tumor cells are checked for this genetic change. The tumor may come back many years after treatment.
- Perivascular epithelioid cell tumors (PEComas). Benign (not cancer) PEComas may be found in children with an inherited condition called tuberous sclerosis. They occur in the stomach, intestines, lungs, female reproductive organs, and genitourinary organs.
- Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor. See the PDQ summary on Ewing Sarcoma Treatment for information.
- Synovial sarcoma. Synovial sarcoma is a common type of soft tissue sarcoma in children and adolescents. Synovial sarcoma usually forms in the tissues around the joints in the arms or legs, but may also form in the trunk, head, or neck. The cells of this tumor usually have a certain genetic change called a translocation (part of one chromosome switches places with part of another chromosome). Larger tumors have a greater risk of spreading to other parts of the body, including the lungs. Children younger than 10 years and those whose tumor is 5 centimeters or smaller have a better prognosis.
- Undifferentiated /unclassified sarcoma. These tumors usually occur in the muscles that are attached to bones and that help the body move.
- Undifferentiated pleomorphic sarcoma /malignant fibrous histiocytoma (high-grade). This type of soft tissue tumor may form in parts of the body where patients have received radiation therapy in the past, or as a second cancer in children with retinoblastoma. The tumor is usually found on the arms or legs and may spread to other parts of the body. See the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for information about malignant fibrous histiocytoma of bone.
Blood vessel tumors
Blood vessel tumors include the following types:
- Angiosarcoma of the soft tissue. Angiosarcoma of the soft tissue is a fast-growing tumor that forms in blood vessels or lymph vessels in any part of the body. Most angiosarcomas are in or just under the skin. Those in deeper soft tissue can form in the liver, spleen, and lung. They are very rare in children, who sometimes have more than one tumor in the skin or liver. Rarely, infantile hemangioma may become angiosarcoma of the soft tissue. (See the PDQ summary on Childhood Vascular Tumors Treatment for more information.)
- Epithelioid hemangioendothelioma. Epithelioid hemangioendotheliomas can occur in children, but are most common in adults between 30 and 50 years of age. They usually occur in the liver, lung, or bone. They may be either fast growing or slow growing. In about a third of cases, the tumor spreads to other parts of the body very quickly. (See the PDQ summary on Childhood Vascular Tumors Treatment for more information.)
See the following PDQ summaries for information about types of soft tissue sarcoma not included in this summary:
- Childhood Rhabdomyosarcoma Treatment.
- Ewing Sarcoma Treatment.
- Unusual Cancers of Childhood Treatment (gastrointestinal stromal tumors).
Certain factors affect prognosis (chance of recovery) and treatment options.
The prognosis (chance of recovery) and treatment options depend on the following:
- The part of the body where the tumor first formed.
- The size and grade of the tumor.
- The type of soft tissue sarcoma.
- How deep the tumor is under the skin.
- Whether the tumor has spread to other places in the body.
- The amount of tumor remaining after surgery to remove it.
- Whether radiation therapy was used to treat the tumor.
- The age and gender of the patient.
- Whether the cancer has just been diagnosed or has recurred (come back).
Stages of Childhood Soft Tissue Sarcoma
After childhood soft tissue sarcoma has been diagnosed, tests are done to find out if cancer cells have spread to other parts of the body.
The process used to find out if cancer has spread within the soft tissue or to other parts of the body is called staging. There is no standard staging system for childhood soft tissue sarcoma.
In order to plan treatment, it is important to know the type of soft tissue sarcoma, whether the tumor can be removed by surgery, and whether cancer has spread to other parts of the body.
The following procedures may be used to find out if cancer has spread:
- Sentinel lymph node biopsy: A sentinel lymph node biopsy is done to check if cancer has spread to the lymph nodes. The sentinel lymph node is the first lymph node to receive lymphatic drainage from a tumor. It is the first lymph node the cancer is likely to spread to from the tumor. A small amount of a radioactive substance and/or blue dye is injected near the tumor. The radioactive substance or dye flows through the lymph ducts to the lymph nodes. The first lymph node to receive the substance or dye is removed. A pathologist views the tissue under a microscope to look for cancer cells. If cancer cells are not found, it may not be necessary to remove more lymph nodes. This procedure is used for epithelioid and clear cell sarcoma.
- PET scan: A PET scan is a procedure to find malignant tumor cells in the body. A small amount of radioactive glucose (sugar) is injected into a vein. The PET scanner rotates around the body and makes a picture of where glucose is being used in the body. Malignant tumor cells show up brighter in the picture because they are more active and take up more glucose than normal cells do. This procedure is also called positron emission tomography (PET) scan.
- PET-CT scan: A procedure that combines the pictures from a PET scan and a computed tomography (CT) scan. The PET and CT scans are done at the same time on the same machine. The pictures from both scans are combined to make a more detailed picture than either test would make by itself.
There are three ways that cancer spreads in the body.
Cancer can spread through tissue, the lymph system, and the blood:
- Tissue. The cancer spreads from where it began by growing into nearby areas.
- Lymph system. The cancer spreads from where it began by getting into the lymph system. The cancer travels through the lymph vessels to other parts of the body.
- Blood. The cancer spreads from where it began by getting into the blood. The cancer travels through the blood vessels to other parts of the body.
Cancer may spread from where it began to other parts of the body.
When cancer spreads to another part of the body, it is called metastasis. Cancer cells break away from where they began (the primary tumor) and travel through the lymph system or blood.
- Lymph system. The cancer gets into the lymph system, travels through the lymph vessels, and forms a tumor (metastatic tumor) in another part of the body.
- Blood. The cancer gets into the blood, travels through the blood vessels, and forms a tumor (metastatic tumor) in another part of the body.
The metastatic tumor is the same type of cancer as the primary tumor. For example, if soft tissue sarcoma spreads to the lung, the cancer cells in the lung are soft tissue sarcoma cells. The disease is metastatic soft tissue sarcoma, not lung cancer.
Recurrent and Progressive Childhood Soft Tissue Sarcoma
Recurrent childhood soft tissue sarcoma is cancer that has recurred (come back) after it has been treated. The cancer may come back in the same place or in other parts of the body.
Progressive childhood soft tissue sarcoma is cancer that did not respond to treatment.
Treatment Option Overview
There are different types of treatment for patients with childhood soft tissue sarcoma.
Different types of treatments are available for patients with childhood soft tissue sarcoma. Some treatments are standard (the currently used treatment), and some are being tested in clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information on new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment.
Because cancer in children is rare, taking part in a clinical trial should be considered. Some clinical trials are open only to patients who have not started treatment.
Children with childhood soft tissue sarcoma should have their treatment planned by a team of health care providers who are experts in treating cancer in children.
Treatment will be overseen by a pediatric oncologist, a doctor who specializes in treating children with cancer. The pediatric oncologist works with other health care providers who are experts in treating children with soft tissue sarcoma and who specialize in certain areas of medicine. These may include a pediatric surgeon with special training in the removal of soft tissue sarcomas. The following specialists may also be included:
- Pediatrician.
- Radiation oncologist.
- Pediatric hematologist.
- Pediatric nurse specialist.
- Rehabilitation specialist.
- Psychologist.
- Social worker.
- Child-life specialist.
Treatment for childhood soft tissue sarcoma may cause side effects.
For information about side effects that begin during treatment for cancer, see our Side Effects page.
Side effects from cancer treatment that begin after treatment and continue for months or years are called late effects. Late effects of cancer treatment may include:
- Physical problems.
- Changes in mood, feelings, thinking, learning, or memory.
- Second cancers (new types of cancer).
Some late effects may be treated or controlled. It is important to talk with your child's doctors about the effects cancer treatment can have on your child. (See the PDQ summary on Late Effects of Treatment for Childhood Cancer for more information.)
Eight types of standard treatment are used:
Surgery
Surgery to completely remove the soft tissue sarcoma is done when possible. If the tumor is very large, radiation therapy or chemotherapy may be given first, to make the tumor smaller and decrease the amount of tissue that needs to be removed during surgery. This is called neoadjuvant therapy.
The following types of surgery may be used:
- Wide local excision: Removal of the tumor along with some normal tissue around it.
- Amputation: Surgery to remove all or part of the limb or appendage with cancer, such as the arm or hand.
- Lymphadenectomy: Removal of the lymph nodes with cancer.
- Mohs surgery: A surgical procedure used to treat cancer in the skin. Individual layers of cancer tissue are removed and checked under a microscope one at a time until all cancer tissue has been removed. This type of surgery is used to treat dermatofibrosarcoma protuberans. It is also called Mohs micrographic surgery.
- Hepatectomy: Surgery to remove all or part of the liver.
A second surgery may be needed to:
- Remove any remaining cancer cells.
- Check the area around where the tumor was removed for cancer cells and then remove more tissue if needed.
If cancer is in the liver, a hepatectomy and liver transplant may be done (the liver is removed and replaced with a healthy one from a donor).
Even if the doctor removes all the cancer that can be seen at the time of the surgery, some patients may be given radiation therapy or chemotherapy after surgery to kill any cancer cells that are left. Treatment given after the surgery, to lower the risk that the cancer will come back, is called adjuvant therapy.
Radiation therapy
Radiation therapy is a cancer treatment that uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. There are two types of radiation therapy:
- External radiation therapy uses a machine outside the body to send radiation toward the cancer. Certain ways of giving radiation therapy can help keep radiation from damaging nearby healthy tissue. This type of radiation therapy may include the following:
- Stereotactic body radiation therapy: Stereotactic body radiation therapy is a type of external radiation therapy. Special equipment is used to place the patient in the same position for each radiation treatment. Once a day for several days, a radiation machine aims a larger than usual dose of radiation directly at the tumor. By having the patient in the same position for each treatment, there is less damage to nearby healthy tissue. This procedure is also called stereotactic external-beam radiation therapy and stereotaxic radiation therapy.
- Internal radiation therapy uses a radioactive substance sealed in needles, seeds, wires, or catheters that are placed directly into or near the cancer.
The way the radiation therapy is given depends on the type and stage of the cancer being treated and whether the tumor was completely removed by surgery. External and internal radiation therapy are used to treat childhood soft tissue sarcoma.
Chemotherapy
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing. When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy). When chemotherapy is placed directly into the cerebrospinal fluid, an organ, or a body cavity such as the abdomen, the drugs mainly affect cancer cells in those areas (regional chemotherapy). Combination chemotherapy is the use of more than one anticancer drug. The way the chemotherapy is given depends on the type of cancer being treated.
Most types of soft tissue sarcoma do not respond to treatment with chemotherapy.
See Drugs Approved for Soft Tissue Sarcoma for more information.
Observation
Observation is closely monitoring a patient's condition without giving any treatment until signs or symptoms appear or change. Observation may be done when:
- Complete removal of the tumor is not possible.
- No other treatments are available.
- The tumor is not likely to damage any vital organs.
Hormone therapy
Hormone therapy is a cancer treatment that removes hormones or blocks their action and stops cancer cells from growing. Hormones are substances made by glands in the body and circulated in the bloodstream. Some hormones can cause certain cancers to grow. If tests show that the cancer cells have places where hormones can attach (receptors), drugs, surgery, or radiation therapy is used to reduce the production of hormones or block them from working. Antiestrogens (drugs that block estrogen), such as tamoxifen, may be used to treat desmoid-type fibromatosis.
Nonsteroidal anti-inflammatory drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are drugs (such as aspirin, ibuprofen, and naproxen) that are commonly used to decrease fever, swelling, pain, and redness. In the treatment of desmoid-type fibromatosis, an NSAID called sulindac may be used to help block the growth of cancer cells.
Targeted therapy
Targeted therapy is a type of treatment that uses drugs or other substances to attack cancer cells. Targeted therapies usually cause less harm to normal cells than chemotherapy or radiation do.
Kinase inhibitors are a type of targeted therapy that block an enzyme called kinase (a type of protein). There are different types of kinases in the body that have different actions.
- ALK inhibitors may stop the cancer from growing and spreading:
- Crizotinib may be used to treat inflammatory myofibroblastic tumor.
- Tyrosine kinase inhibitors (TKIs) block signals needed for tumors to grow:
- Imatinib is used to treat dermatofibrosarcoma protuberans.
- Pazopanib may be used to treat recurrent and progressive soft tissue sarcoma. It is being studied for many types of newly diagnosed soft tissue sarcoma.
- Sorafenib may be used to treat desmoid-type fibromatosis.
New types of tyrosine kinase inhibitors are being studied such as LOXO-101 and entrectinib for infantile fibrosarcoma.
Other types of targeted therapy are being studied in clinical trials, including the following:
- mTOR inhibitors are a type of targeted therapy that stops the protein that helps cells divide and survive. mTOR inhibitors are being studied to treat perivascular epithelioid cell tumors (PEComas) and epithelioid hemangioendothelioma. Sirolimus is a type of mTOR inhibitor therapy.
- Angiogenesis inhibitors are a type of targeted therapy that prevent the growth of new blood vessels needed for tumors to grow. Angiogenesis inhibitors, such as cediranib, sunitinib, and thalidomide are being studied to treat alveolar soft part sarcoma and epithelioid hemangioendothelioma. Bevacizumab is being studied for blood vessel tumors.
- Histone methyltransferase (HMT) inhibitors are targeted therapy drugs that work inside cancer cells and block signals needed for tumors to grow. HMT inhibitors are being studied for the treatment of epithelioid sarcoma, malignant peripheral nerve sheath tumor, extrarenal (extracranial) rhabdoid tumor, extraskeletal myxoid chondrosarcoma, and synovial sarcoma.
- Heat-shock protein inhibitors block certain proteins that protect tumor cells and help them grow. Ganetespib is a heat shock protein inhibitor being studied in combination with the mTOR inhibitor sirolimus for malignant peripheral nerve sheath tumors that cannot be removed by surgery.
- Antibody-drug conjugates are made up of a monoclonal antibody attached to a drug. The monoclonal antibody binds to specific proteins or receptors found on certain cells, including cancer cells. The drug enters these cells and kills them without harming other cells. Lorvotuzumab mertansine is an antibody-drug conjugate being studied for the treatment of rhabdomyosarcoma, malignant peripheral nerve sheath tumor, and synovial sarcoma.
See Drugs Approved for Soft Tissue Sarcoma for more information.
Immunotherapy
Immunotherapy is a treatment that uses the patient's immune system to fight disease. Substances made by the body or made in a laboratory are used to boost, direct, or restore the body's natural defenses against disease.
Interferon is a type of immunotherapy used to treat epithelioid hemangioendothelioma. It interferes with the division of tumor cells and can slow tumor growth.
New types of treatment are being tested in clinical trials.
This summary section describes treatments that are being studied in clinical trials. It may not mention every new treatment being studied. Information about clinical trials is available from the NCI website.
Gene therapy
Gene therapy is being studied for childhood synovial sarcoma that has recurred, spread, or cannot be removed by surgery. Some of the patient's T cells (a type of white blood cell) are removed and the genes in the cells are changed in a laboratory (genetically engineered) so that they will attack specific cancer cells. They are then given back to the patient by infusion.
Patients may want to think about taking part in a clinical trial.
For some patients, taking part in a clinical trial may be the best treatment choice. Clinical trials are part of the cancer research process. Clinical trials are done to find out if new cancer treatments are safe and effective or better than the standard treatment.
Many of today's standard treatments for cancer are based on earlier clinical trials. Patients who take part in a clinical trial may receive the standard treatment or be among the first to receive a new treatment.
Patients who take part in clinical trials also help improve the way cancer will be treated in the future. Even when clinical trials do not lead to effective new treatments, they often answer important questions and help move research forward.
Patients can enter clinical trials before, during, or after starting their cancer treatment.
Some clinical trials only include patients who have not yet received treatment. Other trials test treatments for patients whose cancer has not gotten better. There are also clinical trials that test new ways to stop cancer from recurring (coming back) or reduce the side effects of cancer treatment.
Clinical trials are taking place in many parts of the country. See the Treatment Options section that follows for links to current treatment clinical trials. These have been retrieved from NCI's listing of clinical trials.
Follow-up tests may be needed.
Some of the tests that were done to diagnose the cancer or to find out the stage of the cancer may be repeated. Some tests will be repeated in order to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment may be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your child's condition has changed or if the cancer has recurred (come back). These tests are sometimes called follow-up tests or check-ups.
Treatment Options for Childhood Soft Tissue Sarcoma
For information about the treatments listed below, see the Treatment Option Overview section.
Newly Diagnosed Childhood Soft Tissue Sarcoma
Fat Tissue Tumors
Liposarcoma
Treatment of liposarcoma may include the following:
- Surgery to completely remove the tumor. If the cancer is not completely removed, a second surgery may be done.
- Chemotherapy to shrink the tumor, followed by surgery.
- Radiation therapy before or after surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Bone and Cartilage Tumors
Extraskeletal mesenchymal chondrosarcoma
Treatment of extraskeletal mesenchymal chondrosarcoma may include the following:
- Surgery to completely remove the tumor. Radiation therapy may be given before and/or after surgery.
- Chemotherapy followed by surgery. Chemotherapy with or without radiation therapy is given after surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Extraskeletal osteosarcoma
Treatment of extraskeletal osteosarcoma may include the following:
- Surgery to completely remove the tumor, followed by chemotherapy.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
See the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information.
Fibrous (Connective) Tissue Tumors
Desmoid-type fibromatosis
Treatment of desmoid-type fibromatosis may include the following:
- Surgery to completely remove the tumor.
Treatment before surgery may include the following:
- Observation.
- Chemotherapy.
- Radiation therapy.
- Antiestrogen drug therapy .
- Nonsteroidal anti-inflammatory drug (NSAID) therapy.
If the tumor is not completely removed by surgery, treatment may include the following:
- Observation, if other treatment options are not possible.
- Radiation therapy.
- Radiation therapy or chemotherapy for tumors that cannot be removed by surgery.
- A clinical trial of targeted therapy.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Treatment of desmoid-type fibromatosis that has come back may include the following:
- Observation and possibly surgery at a later time.
- Chemotherapy.
Dermatofibrosarcoma protuberans
Treatment of dermatofibrosarcoma protuberans may include the following:
- Surgery to completely remove the tumor when possible. This may include Mohs surgery.
- Radiation therapy before or after surgery.
- Targeted therapy (imatinib) if the tumor cannot be removed or has come back.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Fibrosarcoma
Infantile fibrosarcoma
Treatment of infantile fibrosarcoma may include the following:
- Surgery to remove the tumor when possible, followed by observation.
- Surgery followed by chemotherapy.
- Chemotherapy to shrink the tumor, followed by surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
- A clinical trial of targeted therapy (tyrosine kinase inhibitor).
Adult-type fibrosarcoma
Treatment of adult-type fibrosarcoma may include the following:
- Surgery to completely remove the tumor when possible.
Inflammatory myofibroblastic tumor
Treatment of inflammatory myofibroblastic tumor may include the following:
- Surgery to completely remove the tumor when possible.
- Chemotherapy.
- Steroid therapy.
- Nonsteroidal anti-inflammatory drug (NSAID) therapy.
- Targeted therapy (ALK inhibitors).
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Low-grade fibromyxoid sarcoma
Treatment of low-grade fibromyxoid sarcoma may include the following:
- Surgery to completely remove the tumor.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Myxofibrosarcoma
Treatment of myxofibrosarcoma may include the following:
- Surgery to completely remove the tumor.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Sclerosing epithelioid fibrosarcoma
Treatment of sclerosing epithelioid fibrosarcoma may include the following:
- Surgery to completely remove the tumor.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Skeletal Muscle Tumors
Rhabdomyosarcoma
See the PDQ summary on Childhood Rhabdomyosarcoma Treatment.
Smooth Muscle Tumors
Leiomyosarcoma
Treatment of leiomyosarcoma may include the following:
- Chemotherapy.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
So-called Fibrohistiocytic Tumors
Plexiform fibrohistiocytic tumor
Treatment of plexiform fibrohistiocytic tumor may include the following:
- Surgery to completely remove the tumor.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Peripheral Nervous System Tumors
Ectomesenchymoma
Treatment of ectomesenchymoma may include the following:
- Surgery and chemotherapy.
- Radiation therapy.
Malignant peripheral nerve sheath tumor
Treatment of malignant peripheral nerve sheath tumor may include the following:
- Surgery to completely remove the tumor when possible.
- Radiation therapy before or after surgery.
- Chemotherapy, for tumors that cannot be removed by surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
- A clinical trial of targeted therapy, for tumors that cannot be removed by surgery.
- A clinical trial of targeted therapy (histone methyltransferase inhibitor).
- A clinical trial of an antibody-drug conjugate.
It is not clear whether giving radiation therapy or chemotherapy after surgery improves the tumor's response to treatment.
Malignant triton tumor
Malignant triton tumors may be treated the same as rhabdomyosarcomas and include surgery, chemotherapy, or radiation therapy. A regimen of targeted therapy, radiation therapy, and surgery with or without chemotherapy is being studied.
Pericytic (Perivascular) Tumors
Infantile hemangiopericytoma
Treatment of infantile hemangiopericytoma may include the following:
Infantile myofibromatosis
Treatment of infantile myofibromatosis may include the following:
- Combination chemotherapy.
Tumors of Unknown Origin (the place where the tumor first formed is not known)
Alveolar soft part sarcoma
Treatment of alveolar soft part sarcoma may include the following:
- Surgery to completely remove the tumor when possible.
- Radiation therapy before or after surgery, if the tumor cannot be completely removed by surgery.
- Targeted therapy (angiogenesis inhibitor).
- A clinical trial of targeted therapy (angiogenesis inhibitor) for children.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Clear cell sarcoma of soft tissue
Treatment of clear cell sarcoma of soft tissue may include the following:
- Surgery to remove the tumor when possible.
- Radiation therapy before or after surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Desmoplastic small round cell tumor
There is no standard treatment for desmoplastic small round cell tumor. Treatment may include the following:
- Surgery to completely remove the tumor when possible.
- Chemotherapy followed by surgery.
- Radiation therapy.
Epithelioid sarcoma
Treatment of epithelioid sarcoma may include the following:
- Surgery to remove the tumor when possible.
- Chemotherapy before or after surgery.
- Radiation therapy before or after surgery.
- A clinical trial of targeted therapy (histone methyltransferase inhibitor).
Extrarenal (extracranial) rhabdoid tumor
Treatment of extrarenal (extracranial) rhabdoid tumor may include the following:
- A combination of surgery to remove the tumor when possible, chemotherapy, and radiation therapy.
- A clinical trial of targeted therapy (histone methyltransferase inhibitor).
Extraskeletal myxoid chondrosarcoma
Treatment of extraskeletal myxoid chondrosarcoma may include the following:
- Surgery to remove the tumor when possible.
- Radiation therapy.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
- A clinical trial of targeted therapy (histone methyltransferase inhibitor).
Perivascular epithelioid cell tumors (PEComas)
Treatment of perivascular epithelioid cell tumors may include the following:
- Surgery to remove the tumor.
- Observation followed by surgery.
- Targeted therapy (mTOR inhibitor), for tumors that have certain gene changes and cannot be removed by surgery.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor
See the PDQ summary on Ewing Sarcoma Treatment.
Synovial sarcoma
Treatment of synovial sarcoma may include the following:
- Chemotherapy.
- Surgery. Radiation therapy and/or chemotherapy may be given before or after surgery.
- A clinical trial of gene therapy.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
- A clinical trial of targeted therapy (histone methyltransferase inhibitor).
- A clinical trial of an antibody-drug conjugate.
Undifferentiated/unclassified sarcoma
These tumors include undifferentiated pleomorphic sarcoma /malignant fibrous histiocytoma (high-grade). There is no standard treatment for these tumors. Treatment may include the following:
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
See the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for information about the treatment of malignant fibrous histiocytoma of bone.
Blood Vessel Tumors
Angiosarcoma of soft tissue
Treatment of angiosarcoma may include the following:
- Surgery to completely remove the tumor.
- A combination of surgery, chemotherapy, and radiation therapy for angiosarcomas that have spread.
- Targeted therapy (bevacizumab) and chemotherapy for angiosarcomas that began as infantile hemangiomas.
- A clinical trial of targeted therapy, radiation therapy, and surgery with or without chemotherapy.
Epithelioid hemangioendothelioma
Treatment of epithelioid hemangioendothelioma may include the following:
- Surgery to remove the tumor when possible.
- Immunotherapy (interferon) and targeted therapy (thalidomide, sorafenib, pazopanib, sirolimus) for tumors that are likely to spread.
- Chemotherapy.
- Total hepatectomy and liver transplant when the tumor is in the liver.
Metastatic Childhood Soft Tissue Sarcoma
Treatment of childhood soft tissue sarcoma that has spread to other parts of the body at diagnosis may include the following:
- Chemotherapy and radiation therapy. Surgery may be done to remove tumors that have spread to the lung.
- Stereotactic body radiation therapy for tumors that have spread to the lung.
For treatment of specific tumor types, see the Treatment Options for Childhood Soft Tissue Sarcoma section.
Check the list of NCI-supported cancer clinical trials that are now accepting patients with nonmetastatic childhood soft tissue sarcoma and metastatic childhood soft tissue sarcoma. For more specific results, refine the search by using other search features, such as the location of the trial, the type of treatment, or the name of the drug. Talk with your child's doctor about clinical trials that may be right for your child. General information about clinical trials is available from the NCI website.
Recurrent and Progressive Childhood Soft Tissue Sarcoma
Treatment of recurrent or progressive childhood soft tissue sarcoma may include the following:
- Surgery to remove cancer that has come back where it first formed or that has spread to the lung.
- Surgery followed by external or internal radiation therapy, if radiation therapy has not already been given.
- Surgery to remove the arm or leg with cancer, if radiation therapy was already given.
- Chemotherapy.
- Targeted therapy (tyrosine kinase inhibitor).
- A clinical trial of a new chemotherapy regimen.
Check the list of NCI-supported cancer clinical trials that are now accepting patients with recurrent childhood soft tissue sarcoma. For more specific results, refine the search by using other search features, such as the location of the trial, the type of treatment, or the name of the drug. Talk with your child's doctor about clinical trials that may be right for your child. General information about clinical trials is available from the NCI website.
To Learn More About Childhood Soft Tissue Sarcoma
For more information from the National Cancer Institute about childhood soft tissue sarcoma, see the following:
- Soft Tissue Sarcoma Home Page
- Computed Tomography (CT) Scans and Cancer
- Drugs Approved for Soft Tissue Sarcoma
- Targeted Cancer Therapies
For more childhood cancer information and other general cancer resources, see the following:
- About Cancer
- Childhood Cancers
- CureSearch for Children's Cancer
- Late Effects of Treatment for Childhood Cancer
- Adolescents and Young Adults with Cancer
- Children with Cancer: A Guide for Parents
- Cancer in Children and Adolescents
- Staging
- Coping with Cancer
- Questions to Ask Your Doctor about Cancer
- For Survivors and Caregivers
About This PDQ Summary
About PDQ
Physician Data Query (PDQ) is the National Cancer Institute's (NCI's) comprehensive cancer information database. The PDQ database contains summaries of the latest published information on cancer prevention, detection, genetics, treatment, supportive care, and complementary and alternative medicine. Most summaries come in two versions. The health professional versions have detailed information written in technical language. The patient versions are written in easy-to-understand, nontechnical language. Both versions have cancer information that is accurate and up to date and most versions are also available in Spanish.
PDQ is a service of the NCI. The NCI is part of the National Institutes of Health (NIH). NIH is the federal government's center of biomedical research. The PDQ summaries are based on an independent review of the medical literature. They are not policy statements of the NCI or the NIH.
Purpose of This Summary
This PDQ cancer information summary has current information about the treatment of childhood soft tissue sarcoma. It is meant to inform and help patients, families, and caregivers. It does not give formal guidelines or recommendations for making decisions about health care.
Reviewers and Updates
Editorial Boards write the PDQ cancer information summaries and keep them up to date. These Boards are made up of experts in cancer treatment and other specialties related to cancer. The summaries are reviewed regularly and changes are made when there is new information. The date on each summary ("Date Last Modified") is the date of the most recent change.
The information in this patient summary was taken from the health professional version, which is reviewed regularly and updated as needed, by the PDQ Pediatric Treatment Editorial Board.
Clinical Trial Information
A clinical trial is a study to answer a scientific question, such as whether one treatment is better than another. Trials are based on past studies and what has been learned in the laboratory. Each trial answers certain scientific questions in order to find new and better ways to help cancer patients. During treatment clinical trials, information is collected about the effects of a new treatment and how well it works. If a clinical trial shows that a new treatment is better than one currently being used, the new treatment may become "standard." Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Clinical trials are listed in PDQ and can be found online at NCI's website. Many cancer doctors who take part in clinical trials are also listed in PDQ. For more information, call the Cancer Information Service 1-800-4-CANCER (1-800-422-6237).
Permission to Use This Summary
PDQ is a registered trademark. The content of PDQ documents can be used freely as text. It cannot be identified as an NCI PDQ cancer information summary unless the whole summary is shown and it is updated regularly. However, a user would be allowed to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks in the following way: [include excerpt from the summary]."
The best way to cite this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/patient/child-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389342]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use in the PDQ summaries only. If you want to use an image from a PDQ summary and you are not using the whole summary, you must get permission from the owner. It cannot be given by the National Cancer Institute. Information about using the images in this summary, along with many other images related to cancer can be found in Visuals Online. Visuals Online is a collection of more than 2,000 scientific images.
Disclaimer
The information in these summaries should not be used to make decisions about insurance reimbursement. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's E-mail Us.
Last Revised: 2017-05-15
If you want to know more about cancer and how it is treated, or if you wish to know about clinical trials for your type of cancer, you can call the NCI's Cancer Information Service at 1-800-422-6237, toll free. A trained information specialist can talk with you and answer your questions.