Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Treatment - Health Professional Information [NCI]Skip to the navigationGeneral Information About Childhood Acute Lymphoblastic Leukemia (ALL)Cancer in children and adolescents is rare, although the overall incidence of childhood cancer, including ALL, has been slowly increasing since 1975.[1] Dramatic improvements in survival have been achieved in children and adolescents with cancer.[1,2,3] Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1,2,3] For ALL, the 5-year survival rate has increased over the same time from 60% to approximately 90% for children younger than 15 years and from 28% to more than 75% for adolescents aged 15 to 19 years.[4] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.) Incidence ALL is the most common cancer diagnosed in children and represents approximately 25% of cancer diagnoses among children younger than 15 years.[2,3] In the United States, ALL occurs at an annual rate of approximately 41 cases per 1 million people aged 0 to 14 years and approximately 17 cases per 1 million people aged 15 to 19 years.[4] There are approximately 3,100 children and adolescents younger than 20 years diagnosed with ALL each year in the United States.[5] Since 1975, there has been a gradual increase in the incidence of ALL.[4,6] A sharp peak in ALL incidence is observed among children aged 2 to 3 years (>90 cases per 1 million per year), with rates decreasing to fewer than 30 cases per 1 million by age 8 years.[2,3] The incidence of ALL among children aged 2 to 3 years is approximately fourfold greater than that for infants and is likewise fourfold to fivefold greater than that for children aged 10 years and older.[2,3] The incidence of ALL appears to be highest in Hispanic children (43 cases per 1 million).[2,3,7,8] The incidence is substantially higher in white children than in black children, with a nearly threefold higher incidence of ALL from age 2 to 3 years in white children than in black children.[2,3,7] Anatomy Childhood ALL originates in the T and B lymphoblasts in the bone marrow (refer to Figure 1). 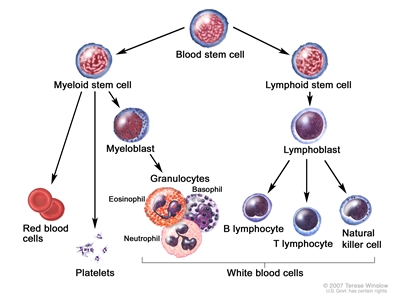
Figure 1. Blood cell development. Different blood and immune cell lineages, including T and B lymphocytes, differentiate from a common blood stem cell.
Marrow involvement of acute leukemia as seen by light microscopy is defined as follows: - M1: Fewer than 5% blast cells.
- M2: 5% to 25% blast cells.
- M3: Greater than 25% blast cells.
Almost all patients with ALL present with an M3 marrow. Risk Factors for Developing ALL Few factors associated with an increased risk of ALL have been identified. The primary accepted risk factors for ALL and associated genes (when relevant) include the following: - Prenatal exposure to x-rays.
- Postnatal exposure to high doses of radiation (e.g., therapeutic radiation as previously used for conditions such as tinea capitis and thymus enlargement).
- Previous treatment with chemotherapy.
- Genetic conditions that include the following:
- Down syndrome. (Refer to the Down syndrome section of this summary for more information.)
- Neurofibromatosis (NF1).[9]
- Bloom syndrome (BLM).[10]
- Fanconi anemia (multiple genes; ALL is observed much less frequently than acute myeloid leukemia [AML]).[11]
- Ataxia telangiectasia (ATM).[12]
- Li-Fraumeni syndrome (TP53).[13,14,15]
- Constitutional mismatch repair deficiency (biallelic mutation of MLH1, MSH2, MSH6, and PMS2).[16,17]
- Low- and high-penetrance inherited genetic variants.[18] (Refer to the Low- and high-penetrance inherited genetic variants section of this summary for more information.)
- Carriers of a constitutional Robertsonian translocation that involves chromosomes 15 and 21 are specifically and highly predisposed to developing iAMP21 ALL.[19]
Down syndrome Children with Down syndrome have an increased risk of developing both ALL and AML,[20,21] with a cumulative risk of developing leukemia of approximately 2.1% by age 5 years and 2.7% by age 30 years.[20,21] Approximately one-half to two-thirds of cases of acute leukemia in children with Down syndrome are ALL, and about 2% to 3% of childhood ALL cases occur in children with Down syndrome.[22,23,24] While the vast majority of cases of AML in children with Down syndrome occur before the age of 4 years (median age, 1 year),[25] ALL in children with Down syndrome has an age distribution similar to that of ALL in non-Down syndrome children, with a median age of 3 to 4 years.[22,23] Patients with ALL and Down syndrome have a lower incidence of both favorable (t(12;21)(p13;q22)/ETV6-RUNX1 [TEL-AML1]) and hyperdiploidy [51-65 chromosomes]) and unfavorable (t(9;22)(q34;q11.2)) or t(4;11)(q21;q23) and hypodiploidy [<44 chromosomes]) cytogenetic findings and a near absence of T-cell phenotype.[22,23,24,25,26] Approximately 50% to 60% of cases of ALL in children with Down syndrome have genomic alterations affecting CRLF2 that generally result in overexpression of the protein produced by this gene, which dimerizes with the interleukin-7 receptor alpha to form the receptor for the cytokine thymic stromal lymphopoietin.[27,28,29]CRLF2 genomic alterations are observed at a much lower frequency (<10%) in children with precursor B-cell ALL who do not have Down syndrome.[29,30,31] Based on the relatively small number of published series, it does not appear that genomic CRLF2 aberrations in patients with Down syndrome and ALL have prognostic relevance.[26,28] However, IKZF1 gene deletions, observed in up to 35% of patients with Down syndrome and ALL, have been associated with a significantly worse outcome in this group of patients.[28,32] Approximately 20% of ALL cases arising in children with Down syndrome have somatically acquired JAK2 mutations,[27,28,33,34,35] a finding that is uncommon among younger children with ALL but that is observed in a subset of primarily older children and adolescents with high-risk precursor B-cell ALL.[36] Almost all Down syndrome ALL cases with JAK2 mutations also have CRLF2 genomic alterations.[27,28,29] Preliminary evidence suggests no correlation between JAK2 mutation status and 5-year event-free survival in children with Down syndrome and ALL,[28,34] but more study is needed to address this issue, as well as the prognostic significance of CRLF2 alterations and IKZF1 gene deletions in this patient population. Low- and high-penetrance inherited genetic variants Genetic predisposition to ALL can be divided into the following several broad categories: - Association with genetic syndromes. Increased risk can be associated with the genetic syndromes listed above in which ALL is observed, although it is not the primary manifestation of the condition.
- Common alleles. Another category for genetic predisposition includes common alleles with relatively small effect sizes that are identified by genome-wide association studies. Genome-wide association studies have identified a number of germline (inherited) genetic polymorphisms that are associated with the development of childhood ALL.[18] For example, the risk alleles of ARID5B are associated with the development of hyperdiploid (51-65 chromosomes) precursor B-cell ALL. ARID5B is a gene that encodes a transcriptional factor important in embryonic development, cell type-specific gene expression, and cell growth regulation.[37,38] Other genes with polymorphisms associated with increased risk of ALL include GATA3,[39]IKZF1,[37,38,40]CDKN2A,[41]CDKN2B,[40,41]CEBPE,[37]PIP4K2A,[39,42] and TP63.[43]
- Rare germline variants with high penetrance. A germline variant in PAX5 that substitutes serine for glycine at amino acid 183 and that reduces PAX5 activity has been identified in several families that experienced multiple cases of ALL.[44,45] Similarly, several germline ETV6 variants that lead to loss of ETV6 function have been identified in kindreds affected by both thrombocytopenia and ALL.[46,47,48] Sequencing of ETV6 in remission (i.e., germline) specimens identified variants that were potentially related to ALL in approximately 1% of children with ALL that were evaluated.[46] This suggests a previously unrecognized contribution to ALL risk that will need to be assessed in future studies.[46,47,48]
Prenatal origin of childhood ALL Development of ALL is in most cases a multistep process, with more than one genomic alteration required for frank leukemia to develop. In at least some cases of childhood ALL, the initial genomic alteration appears to occur in utero. Evidence to support this comes from the observation that the immunoglobulin or T-cell receptor antigen rearrangements that are unique to each patient's leukemia cells can be detected in blood samples obtained at birth.[49,50] Similarly, in ALL characterized by specific chromosomal abnormalities, some patients have blood cells that carry at least one leukemic genomic abnormality at the time of birth, with additional cooperative genomic changes acquired postnatally.[49,50,51] Genomic studies of identical twins with concordant leukemia further support the prenatal origin of some leukemias.[49,52] Evidence also exists that some children who never develop ALL are born with very rare blood cells carrying a genomic alteration associated with ALL. For example, in one study, 1% of neonatal blood spots (Guthrie cards) tested positive for the ETV6-RUNX1 translocation, far exceeding the number of cases of ETV6-RUNX1 ALL in children.[53] Other reports confirm [54] or do not confirm [55,56] this finding, and methodological issues related to fluorescence in situ hybridization testing complicate interpretation of the initial 1% estimate.[57] Clinical Presentation The typical and atypical symptoms and clinical findings of childhood ALL have been published.[58,59,60] Diagnosis The diagnostic evaluation needed to definitively diagnose childhood ALL has been published.[58,59,60,61] The 2016 revision to the World Health Organization classification of tumors of the hematopoietic and lymphoid tissues lists the following entities for acute lymphoid leukemias:[62] B-lymphoblastic leukemia/lymphoma
- B-lymphoblastic leukemia/lymphoma, not otherwise specified (NOS).
- B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities.
- B-lymphoblastic leukemia/lymphoma with t(9;22)(q34.1;q11.2); BCR-ABL1.
- B-lymphoblastic leukemia/lymphoma with t(v;11q23.3); KMT2A rearranged.
- B-lymphoblastic leukemia/lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1.
- B-lymphoblastic leukemia/lymphoma with hyperdiploidy.
- B-lymphoblastic leukemia/lymphoma with hypodiploidy.
- B-lymphoblastic leukemia/lymphoma with t(5;14)(q31.1;q32.3); IL3-IGH.
- B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1.
- Provisional entity: B-lymphoblastic leukemia/lymphoma, BCR-ABL1-like.
- Provisional entity: B-lymphoblastic leukemia/lymphoma with iAMP21.
T-lymphoblastic leukemia/lymphoma
- Provisional entity: Early T-cell precursor lymphoblastic leukemia.
Key clinical and biological characteristics, as well as the prognostic significance for these entities, are discussed in the Cytogenetics/genomics alterations section of this summary. Overall Outcome for ALL Among children with ALL, approximately 98% attain remission, and approximately 85% of patients aged 1 to 18 years with newly diagnosed ALL treated on current regimens are expected to be long-term event-free survivors, with over 90% surviving at 5 years.[63,64,65,66] Despite the treatment advances in childhood ALL, numerous important biologic and therapeutic questions remain to be answered before the goal of curing every child with ALL with the least associated toxicity can be achieved. The systematic investigation of these issues requires large clinical trials, and the opportunity to participate in these trials is offered to most patients and families. Clinical trials for children and adolescents with ALL are generally designed to compare therapy that is currently accepted as standard with investigational regimens that seek to improve cure rates and/or decrease toxicity. In certain trials in which the cure rate for the patient group is very high, therapy reduction questions may be asked. Much of the progress made in identifying curative therapies for childhood ALL and other childhood cancers has been achieved through investigator-driven discovery and tested in carefully randomized, controlled, multi-institutional clinical trials. Information about ongoing clinical trials is available from the NCI website. Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with childhood acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 28. Also available online. Last accessed January 27, 2017.
-
Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 29. Also available online. Last accessed May 31, 2017.
-
Howlader N, Noone AM, Krapcho M: SEER Cancer Statistics Review (CSR) 1975-2013. Bethesda, Md: National Cancer Institute, 2015. Available online. Last accessed January 27, 2017.
-
Special Section: Childhood and Adolescent Cancers. In: American Cancer Society: Cancer Facts and Figures 2014. Atlanta, Ga: American Cancer Society, 2014, pp 25. Available online. Last accessed January 27, 2017.
-
Shah A, Coleman MP: Increasing incidence of childhood leukaemia: a controversy re-examined. Br J Cancer 97 (7): 1009-12, 2007.
-
Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed January 27, 2017.
-
Barrington-Trimis JL, Cockburn M, Metayer C, et al.: Rising rates of acute lymphoblastic leukemia in Hispanic children: trends in incidence from 1992 to 2011. Blood 125 (19): 3033-4, 2015.
-
Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
-
Passarge E: Bloom's syndrome: the German experience. Ann Genet 34 (3-4): 179-97, 1991.
-
Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
-
Taylor AM, Metcalfe JA, Thick J, et al.: Leukemia and lymphoma in ataxia telangiectasia. Blood 87 (2): 423-38, 1996.
-
Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
-
Powell BC, Jiang L, Muzny DM, et al.: Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer 60 (6): E1-3, 2013.
-
Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.
-
Ilencikova D, Sejnova D, Jindrova J, et al.: High-grade brain tumors in siblings with biallelic MSH6 mutations. Pediatr Blood Cancer 57 (6): 1067-70, 2011.
-
Ripperger T, Schlegelberger B: Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 59 (3): 133-42, 2016.
-
Moriyama T, Relling MV, Yang JJ: Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 125 (26): 3988-95, 2015.
-
Li Y, Schwab C, Ryan SL, et al.: Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508 (7494): 98-102, 2014.
-
Hasle H: Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol 2 (7): 429-36, 2001.
-
Whitlock JA: Down syndrome and acute lymphoblastic leukaemia. Br J Haematol 135 (5): 595-602, 2006.
-
Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
-
Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
-
Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
-
Chessells JM, Harrison G, Richards SM, et al.: Down's syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child 85 (4): 321-5, 2001.
-
Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
-
Hertzberg L, Vendramini E, Ganmore I, et al.: Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood 115 (5): 1006-17, 2010.
-
Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
-
Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
-
Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
-
Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
-
Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
-
Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
-
Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
-
Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
-
Mullighan CG, Zhang J, Harvey RC, et al.: JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106 (23): 9414-8, 2009.
-
Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al.: Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1006-10, 2009.
-
Treviño LR, Yang W, French D, et al.: Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1001-5, 2009.
-
Migliorini G, Fiege B, Hosking FJ, et al.: Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122 (19): 3298-307, 2013.
-
Hungate EA, Vora SR, Gamazon ER, et al.: A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun 7: 10635, 2016.
-
Sherborne AL, Hosking FJ, Prasad RB, et al.: Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42 (6): 492-4, 2010.
-
Xu H, Yang W, Perez-Andreu V, et al.: Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105 (10): 733-42, 2013.
-
Ellinghaus E, Stanulla M, Richter G, et al.: Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 26 (5): 902-9, 2012.
-
Shah S, Schrader KA, Waanders E, et al.: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 45 (10): 1226-31, 2013.
-
Auer F, Rüschendorf F, Gombert M, et al.: Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28 (5): 1136-8, 2014.
-
Zhang MY, Churpek JE, Keel SB, et al.: Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 47 (2): 180-5, 2015.
-
Topka S, Vijai J, Walsh MF, et al.: Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet 11 (6): e1005262, 2015.
-
Noetzli L, Lo RW, Lee-Sherick AB, et al.: Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 47 (5): 535-8, 2015.
-
Greaves MF, Wiemels J: Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3 (9): 639-49, 2003.
-
Taub JW, Konrad MA, Ge Y, et al.: High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood 99 (8): 2992-6, 2002.
-
Bateman CM, Colman SM, Chaplin T, et al.: Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115 (17): 3553-8, 2010.
-
Greaves MF, Maia AT, Wiemels JL, et al.: Leukemia in twins: lessons in natural history. Blood 102 (7): 2321-33, 2003.
-
Mori H, Colman SM, Xiao Z, et al.: Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 99 (12): 8242-7, 2002.
-
Zuna J, Madzo J, Krejci O, et al.: ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood 117 (1): 368-9; author reply 370-1, 2011.
-
Lausten-Thomsen U, Madsen HO, Vestergaard TR, et al.: Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood 117 (1): 186-9, 2011.
-
Olsen M, Hjalgrim H, Melbye M, et al.: RT-PCR screening for ETV6-RUNX1-positive clones in cord blood from newborns in the Danish National Birth Cohort. J Pediatr Hematol Oncol 34 (4): 301-3, 2012.
-
Kusk MS, Lausten-Thomsen U, Andersen MK, et al.: False positivity of ETV6/RUNX1 detected by FISH in healthy newborns and adults. Pediatr Blood Cancer 61 (9): 1704-6, 2014.
-
Rabin KR, Gramatges MM, Margolin JF, et al.: Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 463-97.
-
Chessells JM; haemostasis and thrombosis task force, British committee for standards in haematology: Pitfalls in the diagnosis of childhood leukaemia. Br J Haematol 114 (3): 506-11, 2001.
-
Onciu M: Acute lymphoblastic leukemia. Hematol Oncol Clin North Am 23 (4): 655-74, 2009.
-
Heerema-McKenney A, Cleary M, Arber D: Pathology and molecular diagnosis of leukemias and lymphomas. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 113-30.
-
Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
-
Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
-
Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
-
Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
-
Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
Risk-Based Treatment AssignmentIntroduction to Risk-Based Treatment Children with acute lymphoblastic leukemia (ALL) are usually treated according to risk groups defined by both clinical and laboratory features. The intensity of treatment required for favorable outcome varies substantially among subsets of children with ALL. Risk-based treatment assignment is utilized in children with ALL so that patients with favorable clinical and biological features who are likely to have a very good outcome with modest therapy can be spared more intensive and toxic treatment, while a more aggressive, and potentially more toxic, therapeutic approach can be provided for patients who have a lower probability of long-term survival.[1,2] Certain ALL study groups, such as the Children's Oncology Group (COG), use a more- or less-intensive induction regimen based on a subset of pretreatment factors, while other groups give a similar induction regimen to all patients. Factors used by the COG to determine the intensity of induction include immunophenotype, the presence or absence of extramedullary disease, steroid pretreatment, the presence or absence of Down syndrome, and the National Cancer Institute (NCI) risk group classification. The NCI risk group classification for B-cell ALL stratifies risk according to age and white blood cell (WBC) count:[3] - Standard risk-WBC count less than 50,000/μL and age 1 to younger than 10 years.
- High risk-WBC count 50,000/μL or greater and/or age 10 years or older.
All study groups modify the intensity of postinduction therapy based on a variety of prognostic factors, including NCI risk group, immunophenotype, early response determinations, and cytogenetics and genomic alterations.[4] Detection of the Philadelphia chromosome leads to immediate changes in induction therapy.[5] Risk-based treatment assignment requires the availability of prognostic factors that reliably predict outcome. For children with ALL, a number of factors have demonstrated prognostic value, some of which are described below.[6] Factors affecting prognosis are grouped into the following three categories: - Patient and clinical disease characteristics.
- Leukemic characteristics.
- Response to initial treatment.
As in any discussion of prognostic factors, the relative order of significance and the interrelationship of the variables are often treatment dependent and require multivariate analysis to determine which factors operate independently as prognostic variables. Because prognostic factors are treatment dependent, improvements in therapy may diminish or abrogate the significance of any of these presumed prognostic factors. A subset of the prognostic and clinical factors discussed below is used for the initial stratification of children with ALL for treatment assignment. (Refer to the Prognostic (risk) groups under clinical evaluation section of this summary for brief descriptions of the prognostic groupings currently applied in ongoing clinical trials in the United States.) (Refer to the Prognostic Factors After First Relapse of Childhood ALL section of this summary for information about important prognostic factors at relapse.) Prognostic Factors Affecting Risk-Based Treatment Patient and clinical disease characteristics Patient and clinical disease characteristics affecting prognosis include the following: - Age at diagnosis.
- WBC count at diagnosis.
- Central nervous system (CNS) involvement at diagnosis.
- Testicular involvement at diagnosis.
- Down syndrome (trisomy 21).
- Gender.
- Race and ethnicity.
- Weight at diagnosis and during treatment.
Age at diagnosis Age at diagnosis has strong prognostic significance, reflecting the different underlying biology of ALL in different age groups.[7] - Infants (younger than 1 year)
Infants with ALL have a particularly high risk of treatment failure. Treatment failure is most common in the following groups: - Infants younger than 6 months (with an even poorer prognosis for those aged ≤90 days).[8,9,10,11,12]
- Infants with extremely high presenting leukocyte counts (>200,000-300,000 × 109 /L).[9]
- Infants with a poor response to a prednisone prophase.[9]
- Infants with an MLL (KMT2A) gene rearrangement.[8,9,10,11]
Up to 80% of infants with ALL have a translocation of 11q23 with numerous chromosome partners generating an MLL (KMT2A) gene rearrangement.[9,11,13,14] The most common rearrangement is MLL (KMT2A)-AFF1 (t(4;11)(q21;q23)), but MLL rearrangements with many other translocation partners are observed. The rate of MLL (KMT2A) gene rearrangements is extremely high in infants younger than 6 months; from 6 months to 1 year, the incidence of MLL rearrangements decreases but remains higher than that observed in older children.[9,15] Black infants with ALL are significantly less likely to have MLL rearrangements than are white infants.[15] Infants with leukemia and MLL (KMT2A) rearrangements typically have very high WBC counts and an increased incidence of CNS involvement. Event-free survival (EFS) and overall survival (OS) are poor, with 5-year EFS and OS rates of only 35% to 40% for infants with MLL-rearranged ALL.[9,10,11] A comparison of the landscape of somatic mutations in infants and children with MLL-rearranged ALL revealed significant differences between the two groups, suggesting distinctive age-related biological behaviors for MLL-rearranged ALL that may relate to the significantly poorer outcome for infants.[16,17] Blasts from infants with MLL (KMT2A) rearrangements are often CD10 negative and express high levels of FLT3.[9,10,14,18] Conversely, infants whose leukemic cells show a germline MLL gene configuration frequently present with CD10-positive precursor-B immunophenotype. These infants have a significantly better outcome than do infants with ALL characterized by MLL rearrangements.[9,10,14,19] (Refer to the Infants With ALL subsection in the Postinduction Treatment for Specific ALL Subgroups section of this summary for more information about infants with ALL.) - Young children (aged 1 to <10 years)
Young children (aged 1 to <10 years) have a better disease-free survival than older children, adolescents, and infants.[3,7,20,21,22] The improved prognosis in young children is at least partly explained by the more frequent occurrence of favorable cytogenetic features in the leukemic blasts, including hyperdiploidy with 51 to 65 chromosomes and/or favorable chromosome trisomies, or the ETV6-RUNX1 (t(12;21)(p13;q22), also known as the TEL-AML1 translocation).[7,23,24] - Adolescents and young adults (aged ≥10 years)
In general, the outcome of patients aged 10 years and older is inferior to that of patients aged 1 to younger than 10 years. However, the outcome for older children, especially adolescents, has improved significantly over time.[25,26,27] Five-year survival rates for adolescents aged 15 to 19 years increased from 36% (1975-1984) to 72% (2003-2009).[28,29,30] Multiple retrospective studies have established that adolescents aged 16 to 21 years have a better outcome when treated on pediatric versus adult protocols.[31,32,33] (Refer to the Postinduction Treatment for Specific ALL Subgroups section of this summary for more information about adolescents with ALL.)
WBC count at diagnosis A WBC count of 50,000/µL is generally used as an operational cut point between better and poorer prognosis,[3] although the relationship between WBC count and prognosis is a continuous rather than a step function. Patients with precursor B-cell ALL and high WBC counts at diagnosis have an increased risk of treatment failure compared with patients with low initial WBC counts.[34] The median WBC count at diagnosis is much higher for T-cell ALL (>50,000/µL) than for precursor B-cell ALL (<10,000/µL), and there is no consistent effect of WBC count at diagnosis on prognosis for T-cell ALL.[34,35,36,37,38,39,40,41] CNS involvement at diagnosis The presence or absence of CNS leukemia at diagnosis has prognostic significance. Patients who have a nontraumatic diagnostic lumbar puncture may be placed into one of three categories according to the number of WBC/µL and the presence or absence of blasts on cytospin as follows: - CNS1: Cerebrospinal fluid (CSF) that is cytospin negative for blasts regardless of WBC count.
- CNS2: CSF with fewer than 5 WBC/µL and cytospin positive for blasts.
- CNS3 (CNS disease): CSF with 5 or more WBC/µL and cytospin positive for blasts.
Children with ALL who present with CNS disease (CNS3) at diagnosis are at a higher risk of treatment failure (both within the CNS and systemically) than are patients who are classified as CNS1 or CNS2.[42,43] Some studies have reported increased risk of CNS relapse and/or inferior EFS in CNS2 patients, compared with CNS1 patients,[44,45] while others have not.[42,46,47,48] A traumatic lumbar puncture (≥10 erythrocytes/µL) that includes blasts at diagnosis has also been associated with increased risk of CNS relapse and overall poorer outcome in some studies,[42,47,49] but not others.[44,46] Patients with CNS2, CNS3, or traumatic lumbar puncture have a higher frequency of unfavorable prognostic characteristics than do those with CNS1, including significantly higher WBC counts at diagnosis, older age at diagnosis, an increased frequency of the T-cell ALL phenotype, and MLL (KMT2A) gene rearrangements.[42,46,47] Most clinical trial groups have approached CNS2 and traumatic lumbar puncture by utilizing more intensive therapy, primarily additional doses of intrathecal therapy during induction.[42,50,51]; [46][Level of evidence: 2A] To determine whether a patient with a traumatic lumbar puncture (with blasts) should be treated as CNS3, the COG uses an algorithm relating the WBC and red blood cell counts in the spinal fluid and the peripheral blood.[52] Testicular involvement at diagnosis Overt testicular involvement at the time of diagnosis occurs in approximately 2% of males, most commonly in T-cell ALL. In early ALL trials, testicular involvement at diagnosis was an adverse prognostic factor. With more aggressive initial therapy, however, it does not appear that testicular involvement at diagnosis has prognostic significance.[53,54] For example, the European Organization for Research and Treatment of Cancer (EORTC [EORTC-58881]) reported no adverse prognostic significance for overt testicular involvement at diagnosis.[54] The role of radiation therapy for testicular involvement is unclear. A study from St. Jude Children's Research Hospital (SJCRH) suggests that a good outcome can be achieved with aggressive conventional chemotherapy without radiation.[53] The COG has also adopted this strategy for boys with testicular involvement that resolves completely by the end of induction therapy. The COG considers patients with testicular involvement to be high risk regardless of other presenting features, but most other large clinical trial groups in the United States and Europe do not consider testicular disease to be a high-risk feature. Down syndrome (trisomy 21) Outcome in children with Down syndrome and ALL has generally been reported as somewhat inferior to outcomes observed in children who do not have Down syndrome.[55,56,57,58,59] In some studies, the lower EFS and OS of children with Down syndrome appear to be related to increased frequency of treatment-related mortality, as well as higher rates of induction failure and relapse in Down syndrome patients.[55,56,57,58,60,61] The inferior anti-leukemic outcome may be due, in part, to favorable biological features such as ETV6-RUNX1 or hyperdiploidy (51-65 chromosomes) with trisomies of chromosomes 4 and 10 in Down syndrome ALL patients.[60,61] - In a large retrospective study that included 653 patients with Down syndrome and ALL, Down syndrome patients had a lower CR rate (97% vs. 99%, P < .001), higher cumulative incidence of relapse (26% vs. 15%, P < .001) and higher treatment-related mortality (7% vs. < 1%, P < .001) compared with non-Down syndrome patients.[61] Amongst the Down syndrome patients, age younger than 6 years, WBC count of less than 10,000/µL, and the presence of the ETV6-RUNX1 fusion (observed in 8% of patients) were independent predictors of favorable EFS.
- In a report from the COG, among precursor B-cell ALL patients who lacked MLL (KMT2A) rearrangements, BCR-ABL1, ETV6-RUNX1, and hyperdiploidy with trisomies of chromosomes 4 and 10, the EFS and OS were similar in children with and without Down syndrome.[60]
- Certain genomic abnormalities, such as IKZF1 deletions, CRLF2 aberrations, and JAK mutations are seen more frequently in ALL arising in children with Down syndrome than in those without Down syndrome.[62,63,64,65,66] Studies of Down syndrome children with ALL suggest that the presence of IKZF1 deletions (but not CRLF2 aberrations or JAK mutations) is associated with an inferior prognosis.[61,66,67]
Gender In some studies, the prognosis for girls with ALL is slightly better than it is for boys with ALL.[68,69,70] One reason for the better prognosis for girls is the occurrence of testicular relapses among boys, but boys also appear to be at increased risk of bone marrow and CNS relapse for reasons that are not well understood.[68,69,70] While some reports describe outcomes for boys as closely approaching those of girls,[22,50,71] larger clinical trial experiences and national data continue to show somewhat lower survival rates for boys.[21,28,29,72] Race and ethnicity Over the last several decades in the United States, survival rates in black and Hispanic children with ALL have been somewhat lower than the rates in white children with ALL.[73,74] The following factors associated with race and ethnicity influence survival: - ALL subtype. The reason for better outcomes in white and Asian children than in black and Hispanic children is at least partially explained by the different spectrum of ALL subtypes. For example, black children have a higher relative incidence of T-cell ALL and lower rates of favorable genetic subtypes of precursor B-cell ALL.
- Treatment adherence. Differences in outcome may also be related to treatment adherence, as illustrated by two studies of adherence to oral mercaptopurine (6-MP) in maintenance therapy. In the first study, there was an increased risk of relapse in Hispanic children compared with non-Hispanic white children, depending on the level of adherence, even when adjusting for other known variables. However, at adherence rates of 90% or more, Hispanic children continued to demonstrate increased rates of relapse.[75] In the second study, adherence rates were significantly lower in Asian American and African American patients than in non-Hispanic white patients. A greater percentage of patients in these ethnic groups had adherence rates of less than 90%, which was associated with a 3.9-fold increased risk of relapse.[76]
- Ancestry-related genomic variations. Ancestry-related genomic variations may also contribute to racial and ethnic disparities in both the incidence and outcome of ALL.[77] For example, the differential presence of specific host polymorphisms in different racial and ethnic groups may contribute to outcome disparities, as illustrated by the occurrence of single nucleotide polymorphisms in the ARID5B gene that occur more frequently among Hispanics and are linked to both ALL susceptibility and to relapse hazard.[78]
Weight at diagnosis and during treatment Studies of the impact of obesity on the outcome of ALL have had variable results. In most of these studies, obesity is defined as weight above the 95th percentile for age and height. - Three studies have not demonstrated an independent effect of obesity on EFS.[79][Level of evidence: 2Dii]; [80,81][Level of evidence: 3iiDi]
- Two studies have shown obesity to be an independent prognostic factor only in patients older than 10 years or in patients with intermediate-risk or high-risk disease.[82,83][Level of evidence: 3iiDi]
- The COG reported on the impact of obesity on outcome in 2,008 children, 14% of whom were obese, who were enrolled on a high-risk ALL trial (CCG-1961 [NCT00002812]).[84][Level of evidence: 2Di] Obesity was found to be an independent variable for inferior outcome compared with nonobese patients (5-year EFS, 64% vs. 74%; P = .002.) However, obese patients at diagnosis who then normalized their weight during the premaintenance period of treatment had outcomes similar to patients with normal weight at diagnosis.
- In a retrospective study of patients treated at a single institution, obesity at diagnosis was linked to an increased risk of having minimal residual disease (MRD) at the end of induction and an inferior EFS.[85][Level of evidence: 3iiDi]
In a study of 762 pediatric patients with ALL (aged 2-17 years), the Dutch Childhood Oncology Group found that those who were underweight at diagnosis (8% of the population) had an almost twofold higher risk of relapse compared with nonunderweight patients (after adjusting for risk group and age), although this did not result in a difference in EFS or OS. Patients with loss of body mass index during the first 32 weeks of treatment had similar rates of relapse as other patients, but had significantly worse OS, primarily because of poorer salvage rates after relapse.[86] Leukemic characteristics Leukemic cell characteristics affecting prognosis include the following: - Morphology.
- Immunophenotype.
- Cytogenetics/genomic alterations.
Morphology In the past, ALL lymphoblasts were classified using the French-American-British (FAB) criteria as having L1 morphology, L2 morphology, or L3 morphology.[87] However, because of the lack of independent prognostic significance and the subjective nature of this classification system, it is no longer used. Most cases of ALL that show L3 morphology express surface immunoglobulin (Ig) and have a MYC gene translocation identical to those seen in Burkitt lymphoma (i.e., t(8;14)(q24;q32), t(2;8)) that join MYC to one of the immunoglobulin genes. Patients with this specific rare form of leukemia (mature B-cell or Burkitt leukemia) should be treated according to protocols for Burkitt lymphoma. (Refer to the PDQ summary on Childhood Non-Hodgkin Lymphoma Treatment for more information about the treatment of B-cell ALL and Burkitt lymphoma.) Immunophenotype The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia classifies ALL as either B-lymphoblastic leukemia or T-lymphoblastic leukemia, with further subdivisions based on molecular characteristics.[88,89] (Refer to the Diagnosis section of this summary for more information.) Either B- or T-lymphoblastic leukemia can coexpress myeloid antigens. These cases need to be distinguished from leukemia of ambiguous lineage. - Precursor B-cell ALL (WHO B-lymphoblastic leukemia)
Before 2008, the WHO classified B-lymphoblastic leukemia as precursor B-lymphoblastic leukemia, and this terminology is still frequently used in the literature of childhood ALL to distinguish it from mature B-cell ALL. Mature B-cell ALL is now termed Burkitt leukemia and requires different treatment than has been given for precursor B-cell ALL. The older terminology will continue to be used throughout this summary. Precursor B-cell ALL, defined by the expression of cytoplasmic CD79a, CD19, HLA-DR, and other B-cell-associated antigens, accounts for 80% to 85% of childhood ALL. Approximately 90% of precursor B-cell ALL cases express the CD10 surface antigen (formerly known as common ALL antigen [cALLa]). Absence of CD10 is associated with MLL (KMT2A) rearrangements, particularly t(4;11)(q21;q23), and a poor outcome.[9,90] It is not clear whether CD10-negativity has any independent prognostic significance in the absence of an MLL gene rearrangement.[91] The major immunophenotypic subtypes of precursor B-cell ALL are as follows: - Common precursor B-cell ALL (CD10 positive and no surface or cytoplasmic Ig)
Approximately three-quarters of patients with precursor B-cell ALL have the common precursor B-cell immunophenotype and have the best prognosis. Patients with favorable cytogenetics almost always show a common precursor B-cell immunophenotype. - Pro-B ALL (CD10 negative and no surface or cytoplasmic Ig)
Approximately 5% of patients have the pro-B immunophenotype. Pro-B is the most common immunophenotype seen in infants and is often associated with MLL (KMT2A) gene rearrangements. - Pre-B ALL (presence of cytoplasmic Ig)
The leukemic cells of patients with pre-B ALL contain cytoplasmic Ig, and 25% of patients with pre-B ALL have the t(1;19)(q21;p13) translocation with TCF3-PBX1 (previously known as E2A-PBX1) fusion (see below).[92,93] Approximately 3% of patients have transitional pre-B ALL with expression of surface Ig heavy chain without expression of light chain, MYC gene involvement, or L3 morphology. Patients with this phenotype respond well to therapy used for precursor B-cell ALL.[94] Approximately 2% of patients present with mature B-cell leukemia (surface Ig expression, generally with FAB L3 morphology and an 8q24 translocation involving MYC), also called Burkitt leukemia. The treatment for mature B-cell ALL is based on therapy for non-Hodgkin lymphoma and is completely different from that for precursor B-cell ALL. Rare cases of mature B-cell leukemia that lack surface Ig but have L3 morphology with MYC gene translocations should also be treated as mature B-cell leukemia.[94] (Refer to the PDQ summary on Childhood Non-Hodgkin Lymphoma Treatment for more information about the treatment of children with B-cell ALL and Burkitt lymphoma.)
- T-cell ALL
T-cell ALL is defined by expression of the T-cell-associated antigens (cytoplasmic CD3, with CD7 plus CD2 or CD5) on leukemic blasts. T-cell ALL is frequently associated with a constellation of clinical features, including the following:[20,36,71] - Male gender.
- Older age.
- Leukocytosis.
- Mediastinal mass.
With appropriately intensive therapy, children with T-cell ALL have an outcome approaching that of children with B-lineage ALL.[20,36,39,40,71] There are few commonly accepted prognostic factors for patients with T-cell ALL. Conflicting data exist regarding the prognostic significance of presenting leukocyte counts in T-cell ALL.[35,36,37,38,39,40,41,95] The presence or absence of a mediastinal mass at diagnosis has no prognostic significance. In patients with a mediastinal mass, the rate of regression of the mass lacks prognostic significance.[96] Cytogenetic abnormalities common in B-lineage ALL (e.g., hyperdiploidy, 51-65 chromosomes) are rare in T-cell ALL.[97,98] Multiple chromosomal translocations have been identified in T-cell ALL, with structural chromosome rearrangements fusing many genes encoding for transcription factors (e.g., TAL1, LMO1 and LMO2, LYL1, TLX1/HOX11, and TLX3/HOX11L2) to one of the T-cell receptor loci and resulting in aberrant expression of these transcription factors in leukemia cells.[97,99,100,101,102,103] These translocations are often not apparent by examining a standard karyotype, but can be identified using more sensitive screening techniques, such as fluorescence in situ hybridization (FISH) or polymerase chain reaction (PCR).[97] High expression of TLX1/HOX11 resulting from t(10;14)(q24.3;q11.2) involving this gene occurs in 5% to 10% of pediatric T-cell ALL cases and is associated with more favorable outcome in both adults and children with T-cell ALL.[99,100,101,103] Overexpression of TLX3/HOX11L2 resulting from the cryptic t(5;14)(q35;q32) occurs in approximately 20% of pediatric T-cell ALL cases and appears to be associated with increased risk of treatment failure,[101] although not in all studies. Notch pathway signaling is commonly activated by NOTCH1 and FBXW7 gene mutations in T-cell ALL.[104]NOTCH1-activating gene mutations occur in approximately 50% to 60% of T-cell ALL cases, and FBXW7-inactivating gene mutations occur in approximately 15% of cases, with the result that approximately 60% of cases have Notch pathway activation by mutations in at least one of these genes.[105] The prognostic significance of Notch pathway activation by NOTCH1 and FBXW7 mutations in pediatric T-cell ALL is not clear, as some studies have shown a favorable prognosis for mutated cases,[106,107,108,109] while other studies have not shown prognostic significance for the presence of NOTCH1 and/or FBXW7 mutations.[105,110,111,112] A NUP214-ABL1 fusion has been noted in 4% to 6% of T-cell ALL cases and is observed in both adults and children, with a male predominance.[113,114,115] The fusion is cytogenetically cryptic and is seen in FISH on amplified episomes or more rarely, as a small homogeneous staining region.[115] T-cell ALL may also uncommonly show ABL1 fusion proteins with other gene partners (e.g., ETV6, BCR, and EML1).[115]ABL tyrosine kinase inhibitors, such as imatinib or dasatinib, may have therapeutic benefit in this T-cell ALL subtype,[113,114,116] although clinical experience with this strategy is very limited.[117,118,119] Early T-cell precursor ALL Early T-cell precursor ALL, a distinct subset of childhood T-cell ALL, was initially defined by identifying T-cell ALL cases with gene expression profiles highly related to expression profiles for normal early T-cell precursors.[120] The subset of T-cell ALL cases, identified by these analyses represented 13% of all cases and they were characterized by a distinctive immunophenotype (CD1a and CD8 negativity, with weak expression of CD5 and coexpression of stem cell or myeloid markers). Detailed molecular characterization of early T-cell precursor ALL showed this entity to be highly heterogeneous at the molecular level, with no single gene affected by mutation or copy number alteration in more than one-third of cases.[121] Compared with other T-cell ALL cases, the early T-cell precursor group had a lower rate of NOTCH1 mutations and significantly higher frequencies of alterations in genes regulating cytokine receptors and RAS signaling, hematopoietic development, and histone modification. The transcriptional profile of early T-cell precursor ALL shows similarities to that of normal hematopoietic stem cells and myeloid leukemia stem cells.[121] Initial reports describing early T-cell precursor ALL suggested that this subset has a poorer prognosis than other cases of T-cell ALL.[120,122,123] However, another study indicated that the early T-cell precursor ALL subgroup had nonsignificantly inferior 5-year EFS compared with non-early T-cell precursor cases (76% vs. 84%).[124] Similarly, the COG AALL0434 trial observed similar 5-year EFS rates for early T-cell precursor cases and non-early T-cell precursor cases, with both at approximately 87%.[125] Further study in additional patient cohorts is needed to firmly establish the prognostic significance of early T-cell precursor ALL, but most ALL treatment groups do not change patient treatment based on early T-cell precursor status. Studies have found that the absence of biallelic deletion of the TCRgamma locus (ABGD), as detected by comparative genomic hybridization and/or quantitative DNA-PCR, was associated with early treatment failure in patients with T-cell ALL.[126,127] ABGD is characteristic of early thymic precursor cells, and many of the T-cell ALL patients with ABGD have an immunophenotype consistent with the diagnosis of early T-cell precursor phenotype. - Myeloid antigen expression
Up to one-third of childhood ALL cases have leukemia cells that express myeloid-associated surface antigens. Myeloid-associated antigen expression appears to be associated with specific ALL subgroups, notably those with MLL (KMT2A) rearrangements and those with the ETV6-RUNX1 gene rearrangement.[128,129] No independent adverse prognostic significance exists for myeloid-surface antigen expression.[128,129] Leukemia of ambiguous lineage Less than 5% of cases of acute leukemia in children are of ambiguous lineage, expressing features of both myeloid and lymphoid lineage.[130,131,132] These cases are distinct from ALL with myeloid coexpression in that the predominant lineage cannot be determined by immunophenotypic and histochemical studies. The definition of leukemia of ambiguous lineage varies among studies. However, most investigators now use criteria established by the European Group for the Immunological Characterization of Leukemias (EGIL) or the more stringent WHO criteria.[133,134,135] In the WHO classification, the presence of myeloperoxidase is required to establish myeloid lineage. This is not the case for the EGIL classification. Leukemias of mixed phenotype comprise the following two groups:[130] - Bilineal leukemias in which there are two distinct populations of cells, usually one lymphoid and one myeloid.
- Biphenotypic leukemias in which individual blast cells display features of both lymphoid and myeloid lineage. Biphenotypic cases represent the majority of mixed phenotype leukemias.[130] Patients with B-myeloid biphenotypic leukemias lacking the ETV6-RUNX1 fusion have a lower rate of complete remission (CR) and a significantly worse EFS than do patients with precursor B-cell ALL. Some studies suggest that patients with biphenotypic leukemia may fare better with a lymphoid, as opposed to a myeloid, treatment regimen,[131,132,136] although the optimal treatment for patients remains unclear.
Cytogenetics/genomic alterations A number of recurrent chromosomal abnormalities have been shown to have prognostic significance, especially in precursor B-cell ALL. Some chromosomal alterations are associated with more favorable outcomes, such as high hyperdiploidy (51-65 chromosomes) and the ETV6-RUNX1 fusion. Others historically have been associated with a poorer prognosis, including the Philadelphia chromosome (t(9;22)(q34;q11.2)), rearrangements of the MLL (KMT2A) gene, hypodiploidy, and intrachromosomal amplification of the AML1 gene (iAMP21).[137] In recognition of the clinical significance of many of these genomic alterations, the 2016 revision of the World Health Organization classification of tumors of the hematopoietic and lymphoid tissues lists the following entities for precursor B-cell ALL:[89] - B-lymphoblastic leukemia/lymphoma, not otherwise specified (NOS).
- B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities.
- B-lymphoblastic leukemia/lymphoma with t(9;22)(q34.1;q11.2); BCR-ABL1.
- B-lymphoblastic leukemia/lymphoma with t(v;11q23.3); KMT2A rearranged.
- B-lymphoblastic leukemia/lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1.
- B-lymphoblastic leukemia/lymphoma with hyperdiploidy.
- B-lymphoblastic leukemia/lymphoma with hypodiploidy.
- B-lymphoblastic leukemia/lymphoma with t(5;14)(q31.1;q32.3); IL3-IGH.
- B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1.
- Provisional entity: B-lymphoblastic leukemia/lymphoma, BCR-ABL1-like.
- Provisional entity: B-lymphoblastic leukemia/lymphoma with iAMP21.
These and other chromosomal and genomic abnormalities for childhood ALL are described below. - Chromosome number
- High hyperdiploidy (51-65 chromosomes)
High hyperdiploidy, defined as 51 to 65 chromosomes per cell or a DNA index greater than 1.16, occurs in 20% to 25% of cases of precursor B-cell ALL, but very rarely in cases of T-cell ALL.[138] Hyperdiploidy can be evaluated by measuring the DNA content of cells (DNA index) or by karyotyping. In cases with a normal karyotype or in which standard cytogenetic analysis was unsuccessful, interphase fluorescence in situ hybridization (FISH) may detect hidden hyperdiploidy. High hyperdiploidy generally occurs in cases with clinically favorable prognostic factors (patients aged 1 to <10 years with a low white blood cell [WBC] count) and is itself an independent favorable prognostic factor.[24,138,139] Within the hyperdiploid range of 51 to 65 chromosomes, patients with higher modal numbers (58-66) appeared to have a better prognosis in one study.[24] Hyperdiploid leukemia cells are particularly susceptible to undergoing apoptosis and accumulate higher levels of methotrexate and its active polyglutamate metabolites,[140] which may explain the favorable outcome commonly observed in these cases. While the overall outcome of patients with high hyperdiploidy is considered to be favorable, factors such as age, WBC count, specific trisomies, and early response to treatment have been shown to modify its prognostic significance.[141,142] Patients with trisomies of chromosomes 4, 10, and 17 (triple trisomies) have been shown to have a particularly favorable outcome as demonstrated by both Pediatric Oncology Group (POG) and Children's Cancer Group analyses of NCI standard-risk ALL.[143] POG data suggest that NCI standard-risk patients with trisomies of 4 and 10, without regard to chromosome 17 status, have an excellent prognosis.[144] Chromosomal translocations may be seen with high hyperdiploidy, and in those cases, patients are more appropriately risk-classified based on the prognostic significance of the translocation. For instance, in one study, 8% of patients with the Philadelphia chromosome (t(9;22)(q34;q11.2)) also had high hyperdiploidy,[145] and the outcome of these patients (treated without tyrosine kinase inhibitors) was inferior to that observed in non-Philadelphia chromosome-positive (Ph+) high hyperdiploid patients. Certain patients with hyperdiploid ALL may have a hypodiploid clone that has doubled (masked hypodiploidy).[146] These cases may be interpretable based on the pattern of gains and losses of specific chromosomes. These patients have an unfavorable outcome, similar to those with hypodiploidy.[146] Near triploidy (68-80 chromosomes) and near tetraploidy (>80 chromosomes) are much less common and appear to be biologically distinct from high hyperdiploidy.[147] Unlike high hyperdiploidy, a high proportion of near tetraploid cases harbor a cryptic ETV6-RUNX1 fusion.[147,148,149] Near triploidy and tetraploidy were previously thought to be associated with an unfavorable prognosis, but later studies suggest that this may not be the case.[147,149] The genomic landscape of hyperdiploid ALL is represented by mutations in genes of the receptor tyrosine kinase (RTK)/RAS pathway in approximately one-half of cases. Genes encoding histone modifiers are also present in a recurring manner in a minority of cases. Analysis of mutation profiles demonstrates that chromosomal gains are early events in the pathogenesis of hyperdiploid ALL.[150] - Hypodiploidy (<44 chromosomes)
Precursor B-cell ALL cases with fewer than the normal number of chromosomes have been subdivided in various ways, with one report stratifying based on modal chromosome number into the following four groups:[146] - Near-haploid: 24 to 29 chromosomes (n = 46).
- Low-hypodiploid: 33 to 39 chromosomes (n = 26).
- High-hypodiploid: 40 to 43 chromosomes (n = 13).
- Near-diploid: 44 chromosomes (n = 54).
Most patients with hypodiploidy are in the near-haploid and low-hypodiploid groups, and both of these groups have an elevated risk of treatment failure compared with nonhypodiploid cases.[146,151] Patients with fewer than 44 chromosomes have a worse outcome than do patients with 44 or 45 chromosomes in their leukemic cells.[146] One study of 20 patients with near-haploid or low-hypodiploid ALL indicated that MRD may have prognostic significance in the hypodiploid population.[152] The recurring genomic alterations of near-haploid and low-hypodiploid ALL appear to be distinctive from each other and from other types of ALL.[153] In near-haploid ALL, alterations targeting RTK signaling, RAS signaling, and IKZF3 are common.[154] In low-hypodiploid ALL, genetic alterations involving TP53, RB1, and IKZF2 are common. Importantly, the TP53 alterations observed in low-hypodiploid ALL are also present in nontumor cells in approximately 40% of cases, suggesting that these mutations are germline and that low-hypodiploid ALL represents, in some cases, a manifestation of Li-Fraumeni syndrome.[153]
- Chromosomal translocations and gains/deletions of chromosomal segments
- t(12;21)(p13.2;q22.1); ETV6-RUNX1 (formerly known as TEL-AML1)
Fusion of the ETV6 gene on chromosome 12 to the RUNX1 gene on chromosome 21 is present in 20% to 25% of cases of precursor B-cell ALL but is rarely observed in T-cell ALL.[148] The t(12;21)(p12;q22) produces a cryptic translocation that is detected by methods such as FISH, rather than conventional cytogenetics, and it occurs most commonly in children aged 2 to 9 years.[155,156] Hispanic children with ALL have a lower incidence of t(12;21)(p13;q22) than do white children.[157] Reports generally indicate favorable event-free survival (EFS) and overall survival (OS) in children with the ETV6-RUNX1 fusion; however, the prognostic impact of this genetic feature is modified by the following factors:[158,159,160,161,162] - Early response to treatment.
- NCI risk category (age and WBC count at diagnosis).
- Treatment regimen.
In one study of the treatment of newly diagnosed children with ALL, multivariate analysis of prognostic factors found age and leukocyte count, but not ETV6-RUNX1, to be independent prognostic factors.[158] It does not appear that the presence of secondary cytogenetic abnormalities, such as deletion of ETV6 (12p) or CDKN2A/B (9p), impacts the outcome of patients with the ETV6-RUNX1 fusion.[162,163] There is a higher frequency of late relapses in patients with ETV6-RUNX1 fusion compared with other precursor B-cell ALL.[158,164] Patients with the ETV6-RUNX1 fusion who relapse seem to have a better outcome than other relapse patients,[165] with an especially favorable prognosis for patients who relapse more than 36 months from diagnosis.[166] Some relapses in patients with t(12;21)(p13;q22) may represent a new independent second hit in a persistent preleukemic clone (with the first hit being the ETV6-RUNX1 translocation).[167,168] - t(9;22)(q34.1;q11.2); BCR-ABL1 (Ph+)
The Philadelphia chromosome t(9;22)(q34;q11.2) is present in approximately 3% of children with ALL and leads to production of a BCR-ABL1 fusion protein with tyrosine kinase activity (refer to Figure 2).
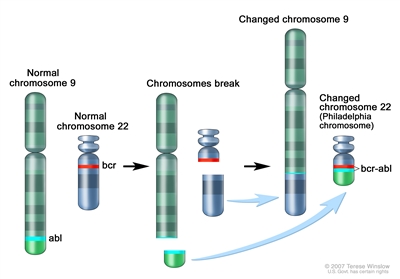
Figure 2. The Philadelphia chromosome is a translocation between the ABL-1 oncogene (on the long arm of chromosome 9) and the breakpoint cluster region (BCR) (on the long arm of chromosome 22), resulting in the fusion gene BCR-ABL1. BCR-ABL1 encodes an oncogenic protein with tyrosine kinase activity. This subtype of ALL is more common in older children with precursor B-cell ALL and high WBC count, with the incidence of the t(9;22)(q34;q11.2) increasing to about 25% in young adults with ALL. Historically, the Philadelphia chromosome t(9;22)(q34;q11.2) was associated with an extremely poor prognosis (especially in those who presented with a high WBC count or had a slow early response to initial therapy), and its presence had been considered an indication for allogeneic hematopoietic stem cell transplantation (HSCT) in patients in first remission.[145,169,170,171] Inhibitors of the BCR-ABL1 tyrosine kinase, such as imatinib mesylate, are effective in patients with Ph+ ALL.[172] A study by the Children's Oncology Group (COG), which used intensive chemotherapy and concurrent imatinib mesylate given daily, demonstrated a 5-year EFS rate of 70% ± 12%, which was superior to the EFS rate of historical controls in the pre-tyrosine kinase inhibitor (imatinib mesylate) era.[173,174] - t(v;11q23.3); MLL (KMT2A)-rearranged
Rearrangements involving the MLL (KMT2A) gene occur in approximately 5% of childhood ALL cases overall, but in up to 80% of infants with ALL. These rearrangements are generally associated with an increased risk of treatment failure.[90,175,176,177] The t(4;11)(q21;q23) is the most common rearrangement involving the MLL gene in children with ALL and occurs in approximately 1% to 2% of childhood ALL.[175,178] Patients with the t(4;11)(q21;q23) are usually infants with high WBC counts; they are more likely than other children with ALL to have central nervous system (CNS) disease and to have a poor response to initial therapy.[9] While both infants and adults with the t(4;11)(q21;q23) are at high risk of treatment failure, children with the t(4;11)(q21;q23) appear to have a better outcome than either infants or adults.[90,175] Irrespective of the type of MLL (KMT2A) gene rearrangement, infants with leukemia cells that have MLL gene rearrangements have a worse treatment outcome than older patients whose leukemia cells have an MLL gene rearrangement.[90,175] Whole-genome sequencing has determined that cases of infant ALL with MLL gene rearrangements have few additional genomic alterations, none of which have clear clinical significance.[17] Deletion of the MLL gene has not been associated with an adverse prognosis.[179] Of interest, the t(11;19)(q23;p13.3) involving MLL (KMT2A) and MLLT1/ENL occurs in approximately 1% of ALL cases and occurs in both early B-lineage and T-cell ALL.[180] Outcome for infants with the t(11;19) is poor, but outcome appears relatively favorable in older children with T-cell ALL and the t(11;19).[180] - t(1;19)(q23;p13.3); TCF3-PBX1 and t(17;19)(q22;p13); TCF3-HLF
The t(1;19) occurs in approximately 5% of childhood ALL cases and involves fusion of the TCF3 gene on chromosome 19 to the PBX1 gene on chromosome 1.[92,93] The t(1;19) may occur as either a balanced translocation or as an unbalanced translocation and is the primary recurring genomic alteration of the pre-B ALL immunophenotype (cytoplasmic Ig positive).[181] Black children are relatively more likely than white children to have pre-B ALL with the t(1;19).[182] The t(1;19) had been associated with inferior outcome in the context of antimetabolite-based therapy,[183] but the adverse prognostic significance was largely negated by more aggressive multiagent therapies.[93,184] However, in a trial conducted by St. Jude Children's Research Hospital (SJCRH) on which all patients were treated without cranial radiation, patients with the t(1;19) had an overall outcome comparable to children lacking this translocation, with a higher risk of CNS relapse and a lower rate of bone marrow relapse, suggesting that more intensive CNS therapy may be needed for these patients.[50,185] The t(17;19) resulting in the TCF3-HLF fusion occurs in less than 1% of pediatric ALL cases. ALL with the TCF3-HLF fusion is associated at diagnosis with disseminated intravascular coagulation and with hypercalcemia. Outcome is very poor for children with the t(17;19), with a literature review noting mortality for 20 of 21 cases reported.[186] In addition to the TCF3-HLF fusion, the genomic landscape of this ALL subtype was characterized by deletions in genes involved in B-cell development (PAX5, BTG1, and VPREB1) and by mutations in RAS pathway genes (NRAS, KRAS, and PTPN11).[181] - DUX4-rearranged ALL with frequent ERG deletions
Approximately 5% of standard-risk and 10% of high-risk pediatric precursor B-cell ALL patients have a rearrangement involving DUX4 that leads to its overexpression.[187,188] The frequency in older adolescents (aged >15 years) is approximately 10%. The most common rearrangement produces IGH-DUX4 fusions, with ERG-DUX4 fusions also observed. Approximately 50% of DUX4-rearranged cases have focal intragenic deletions involving ERG that are not observed in other ALL subtypes,[187,188] and DUX4-rearranged cases show a distinctive gene expression pattern that was initially identified as being associated with these focal deletions in ERG.[189,190,191]IKZF1 alterations are observed in 35% to 40% of DUX4-rearranged ALL.[187,188]ERG deletion connotes an excellent prognosis, with OS exceeding 90%; even when the IZKF1 deletion is present, prognosis remains highly favorable.[189,190,191] Patients with DUX4 rearrangements who lack ERG deletion also appear to have favorable prognosis.[188] - t(5;14)(q31.1;q32.3); IL3-IGH
This entity is included in the 2016 revision of the WHO classification of tumors of the hematopoietic and lymphoid tissues.[89] The finding of t(5;14)(q31.1;q32.3) in patients with ALL and hypereosinophilia in the 1980s was followed by the identification of the IL3-IGH fusion as the underlying genetic basis for the condition.[192,193] The joining of the IGH locus to the promoter region of the interleukin-3 gene (IL3) leads to dysregulation of IL3 expression.[194] Cytogenetic abnormalities in children with ALL and eosinophilia are variable, with only a subset resulting from the IL3-IGH fusion.[195] The number of cases of IL3-IGH ALL described in the published literature is too small to assess the prognostic significance of the IL3-IGH fusion. - Intrachromosomal amplification of chromosome 21 (iAMP21)
iAMP21 with multiple extra copies of the RUNX1 (AML1) gene at 21q22 occurs in approximately 2% of precursor B-cell ALL cases and is associated with older age (median, approximately 10 years), presenting WBC of less than 50 × 109 /L, a slight female preponderance, and high end-induction minimal residual disease (MRD).[196,197,198] The United Kingdom (UK)-ALL clinical trials group initially reported that the presence of iAMP21 conferred a poor prognosis in patients treated in the MRC ALL 97/99 trial (5-year EFS, 29%).[137] In their subsequent trial (UKALL2003 [NCT00222612]), patients with iAMP21 were assigned to a more intensive chemotherapy regimen and had a markedly better outcome (5-year EFS, 78%).[197] Similarly, the COG has reported that iAMP21 was associated with a significantly inferior outcome in NCI standard-risk patients (4-year EFS, 73% for iAMP21 vs. 92% in others), but not in NCI high-risk patients (4-year EFS, 73% vs. 80%).[196] On multivariate analysis, iAMP21 was an independent predictor of inferior outcome only in NCI standard-risk patients.[196] The results of the UKALL2003 and COG studies suggest that treatment of iAMP21 patients with high-risk chemotherapy regimens abrogates its adverse prognostic significance and obviates the need for SCT in first remission.[198] - IKZF1 deletions
IKZF1 deletions, including deletions of the entire gene and deletions of specific exons, are present in approximately 15% of precursor B-cell ALL cases. Less commonly, IKZF1 can be inactivated by deleterious point mutations.[199] Cases with IKZF1 deletions tend to occur in older children, have a higher WBC count at diagnosis, and are therefore, more common in NCI high-risk patients than in NCI standard-risk patients.[199,200,201,202] A high proportion of BCR-ABL1 cases have a deletion of IKZF1,[201,203] and ALL arising in children with Down syndrome appears to have elevated rates of IKZF1 deletions.[66]IKZF1 deletions are also common in cases with CRLF2 genomic alterations and in Philadelphia chromosome (Ph)-like (BCR-ABL1-like) ALL (see below).[189,201,204] Multiple reports have documented the adverse prognostic significance of an IKZF1 deletion, and most studies have reported that this deletion is an independent predictor of poor outcome on multivariate analyses.[189,199,201,204,205,206,207,208,209,210]; [211][Level of evidence: 2Di] That said, the prognostic significance of IKZF1 may not apply equally across ALL biological subtypes, as illustrated by the apparent lack of prognostic significance in patients with ERG deletion.[190] - BCR-ABL1-like (Ph-like)
BCR-ABL1-negative patients with a gene expression profile similar to BCR-ABL1-positive patients have been referred to as BCR-ABL1-like.[199,204] This occurs in 10% to 15% of pediatric ALL patients, increasing in frequency with age, and has been associated with a poor prognosis and with IKZF1 deletion or mutation.[199,204,208,212,213] The 5-year EFS rate of 90% observed in one study of 40 patients with BCR-ABL1-like ALL suggested that the adverse prognostic significance of this subtype may be abrogated when patients are treated with risk-directed therapy based on MRD levels. Six of these 40 patients were classified as high risk and all proceeded to allogeneic SCT.[214][Level of evidence: 2A] The hallmark of BCR-ABL1-like ALL is activated kinase signaling, with 50% containing CRLF2 genomic alterations [215] and half of those cases containing concomitant JAK mutations.[62] Additional information about BCR-ABL1-like ALL cases with CRLF2 genomic alterations is provided below. Many of the remaining cases of BCR-ABL1-like ALL have been noted to have a series of translocations with a common theme of involvement of kinases, including ABL1, ABL2, CSF1R, JAK2, and PDGFRB.[212,216] Fusion proteins from these gene combinations have been noted in some cases to be transformative and have responded to tyrosine kinase inhibitors both in vitro and in vivo,[212] suggesting potential therapeutic strategies for these patients. Point mutations in kinase genes, aside from those in JAK1 and JAK2, are uncommon in Ph-like ALL cases.[213] Genomic alterations in CRLF2, a cytokine receptor gene located on the pseudoautosomal regions of the sex chromosomes, have been identified in 5% to 10% of cases of precursor B-cell ALL; they represent approximately 50% of cases of BCR-ABL1-like ALL.[217,218] The chromosomal abnormalities that commonly lead to CRLF2 overexpression include translocations of the IgH locus (chromosome 14) to CRLF2 and interstitial deletions in pseudoautosomal regions of the sex chromosomes, resulting in a P2RY8-CRLF2 fusion.[213,215,217,218]CRLF2 abnormalities are strongly associated with the presence of IKZF1 deletions and JAK mutations;[62,201,213,215,218] they are also more common in children with Down syndrome.[218] Point mutations in tyrosine kinase genes other than JAK1 and JAK2 are uncommon in CRLF2-overexpressing cases.[213] Although the results of several retrospective studies suggest that CRLF2 abnormalities may have adverse prognostic significance on univariate analyses, most do not find this abnormality to be an independent predictor of outcome.[215,217,218,219,220] For example, in a large European study, increased expression of CRLF2 was not associated with unfavorable outcome in multivariate analysis, while IKZF1 deletion and BCR-ABL1-like expression signatures were associated with unfavorable outcome.[208] Controversy exists about whether the prognostic significance of CRLF2 abnormalities should be analyzed based on CRLF2 overexpression or on the presence of CRLF2 genomic alterations.[219,220] Approximately 9% of BCR-ABL1-like ALL cases result from rearrangements that lead to overexpression of a truncated erythropoietin receptor (EPOR).[221] The C-terminal region of the receptor that is lost is the region that is mutated in primary familial congenital polycythemia and that controls stability of the EPOR. The portion of the EPOR remaining is sufficient for JAK-STAT activation and for driving leukemia development.
- Gene polymorphisms in drug metabolic pathways
A number of polymorphisms of genes involved in the metabolism of chemotherapeutic agents have been reported to have prognostic significance in childhood ALL.[222,223,224] For example, patients with mutant phenotypes of thiopurine methyltransferase (TPMT, a gene involved in the metabolism of thiopurines, such as mercaptopurine [6-MP]), appear to have more favorable outcomes,[225] although such patients may also be at higher risk of developing significant treatment-related toxicities, including myelosuppression and infection.[226,227] Patients with homozygosity for TPMT variants associated with low enzymatic activity tolerate only very low doses of mercaptopurine (approximately 10% of the standard dose) and are treated with reduced doses of mercaptopurine to avoid excessive toxicity. Patients who are heterozygous for this mutant enzyme gene generally tolerate mercaptopurine without serious toxicity, but they do require more frequent dose reductions for hematologic toxicity than do patients who are homozygous for the normal allele.[228,229] Germline variants in nucleoside diphosphate-linked moiety X-type motif 15 (NUDT15) that reduce or abolish activity of this enzyme also lead to diminished tolerance to thiopurines.[228,230] The variants are most common in East Asians and Hispanics, and they are rare in Europeans and Africans. Patients homozygous for the risk variants tolerate only very low doses of mercaptopurine, while patients heterozygous for the risk alleles tolerate lower doses than do patients homozygous for the wild-type allele (approximately 25% dose reduction on average), but there is broad overlap in tolerated doses between the two groups.[228,231] Gene polymorphisms may also affect the expression of proteins that play central roles in the cellular effects of anticancer drugs. As an example, patients who are homozygous for a polymorphism in the promoter region of CEP72 (a centrosomal protein involved in microtubule formation) are at increased risk of vincristine neurotoxicity.[232] Genome-wide polymorphism analysis has identified specific single nucleotide polymorphisms associated with high end-induction MRD and risk of relapse. Polymorphisms of IL-15, as well as genes associated with the metabolism of etoposide and methotrexate, were significantly associated with treatment response in two large cohorts of ALL patients treated on SJCRH and COG protocols.[233] Polymorphic variants involving the reduced folate carrier and methotrexate metabolism have been linked to toxicity and outcome.[234,235] While these associations suggest that individual variations in drug metabolism can affect outcome, few studies have attempted to adjust for these variations; it is unknown whether individualized dose modification based on these findings will improve outcome.
Response to initial treatment The rapidity with which leukemia cells are eliminated after initiation of treatment and the level of residual disease at the end of induction are associated with long-term outcome. Because treatment response is influenced by the drug sensitivity of leukemic cells and host pharmacodynamics and pharmacogenomics,[236] early response has strong prognostic significance. Various ways of evaluating the leukemia cell response to treatment have been utilized, including the following: - MRD determination.
- Day 7 and day 14 bone marrow responses.
- Peripheral blood response to steroid prophase.
- Peripheral blood response to multiagent induction therapy.
- Peripheral blood MRD before end of induction (day 8, day 15).
- Marrow morphology at the end of induction (induction failure).
MRD determination Morphologic assessment of residual leukemia in blood or bone marrow is often difficult and is relatively insensitive. Traditionally, a cutoff of 5% blasts in the bone marrow (detected by light microscopy) has been used to determine remission status. This corresponds to a level of 1 in 20 malignant cells. If one wishes to detect lower levels of leukemic cells in either blood or marrow, specialized techniques such as PCR assays, which determine unique Ig/T-cell receptor gene rearrangements, fusion transcripts produced by chromosome translocations, or flow cytometric assays, which detect leukemia-specific immunophenotypes, are required. With these techniques, detection of as few as 1 leukemia cell in 100,000 normal cells is possible, and MRD at the level of 1 in 10,000 cells can be detected routinely.[237] Multiple studies have demonstrated that end-induction MRD is an important, independent predictor of outcome in children and adolescents with B-lineage ALL.[159,238,239] MRD response discriminates outcome in subsets of patients defined by age, leukocyte count, and cytogenetic abnormalities.[240] Patients with higher levels of end-induction MRD have a poorer prognosis than those with lower or undetectable levels.[159,237,238,239] End-induction MRD is used by almost all groups as a factor determining the intensity of postinduction treatment, with patients found to have higher levels allocated to more intensive therapies. MRD levels at earlier time points during induction (e.g., day 8 and day 15) and later time points (e.g., week 12 of therapy) also predict outcome.[159,237,241]; [242][Level of evidence: 2A] MRD levels obtained 10 to 12 weeks after the start of treatment (end-consolidation) have also been shown to be prognostically important; patients with high levels of MRD at this time point have a significantly inferior EFS compared with other patients.[239,240] - B-cell ALL: For patients with B-cell ALL, evaluating MRD at two time points (end-induction and end-consolidation) can identify the following three prognostically distinct patient subsets:[240]
- Low or undetectable end-induction MRD-best prognosis.
- Detectable or high MRD at end-induction but low or negative end-consolidation MRD-intermediate prognosis.
- Detectable or high MRD at end-consolidation (week 12 of therapy)-worst prognosis.
- T-cell ALL: There are fewer studies documenting the prognostic significance of MRD in patients with T-cell ALL. In the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) ALL-BFM-2000 (NCT00430118) trial, MRD status at day 78 (week 12) was the most important predictor for relapse in patients with T-cell ALL.[243] Patients with detectable MRD at end-induction who had negative MRD by day 78 generally had a favorable prognosis similar to that of patients who achieved MRD-negativity at the earlier end-induction time point.[243]
MRD measurements, in conjunction with other presenting features, have also been used to identify subsets of patients with an extremely low risk of relapse. The COG reported a very favorable prognosis (5-year EFS of 97% ± 1%) for patients with precursor B-cell phenotype, NCI standard risk age/leukocyte count, CNS1 status, and favorable cytogenetic abnormalities (either high hyperdiploidy with favorable trisomies or the ETV6-RUNX1 fusion) who had less than 0.01% MRD levels at both day 8 (from peripheral blood) and end-induction (from bone marrow).[159] The excellent outcomes in patients with low MRD at the end of induction is sustained for more than 10 years from diagnosis.[244] Modifying therapy based on MRD determination has been shown to improve outcome. Day 7 and day 14 bone marrow responses Patients who have a rapid reduction in leukemia cells to less than 5% in their bone marrow within 7 or 14 days after the initiation of multiagent chemotherapy have a more favorable prognosis than do patients who have slower clearance of leukemia cells from the bone marrow.[247] MRD assessments at the end of induction therapy have generally replaced day 7 and day 14 morphological assessments as response to therapy prognostic indicators because the latter lose their prognostic significance in multivariate analysis once MRD is included in the analyses.[159,248] Peripheral blood response to steroid prophase Patients with a reduction in peripheral blast count to less than 1,000/µL after a 7-day induction prophase with prednisone and one dose of intrathecal methotrexate (a good prednisone response) have a more favorable prognosis than do patients whose peripheral blast counts remain above 1,000/µL (a poor prednisone response).[20] Poor prednisone response is observed in fewer than 10% of patients.[20,249] Treatment stratification for protocols of the Berlin-Frankfurt-Münster (BFM) clinical trials group is partially based on early response to the 7-day prednisone prophase (administered immediately before the initiation of multiagent remission induction). Peripheral blood response to multiagent induction therapy Patients with persistent circulating leukemia cells at 7 to 10 days after the initiation of multiagent chemotherapy are at increased risk of relapse compared with patients who have clearance of peripheral blasts within 1 week of therapy initiation.[250] Rate of clearance of peripheral blasts has been found to be of prognostic significance in both T-cell and B-lineage ALL.[250] Peripheral blood MRD before end of induction (day 8, day 15) MRD using peripheral blood obtained 1 week after the initiation of multiagent induction chemotherapy has also been evaluated as an early response-to-therapy prognostic factor. - In a COG study involving nearly 2,000 children with ALL, the presence of MRD in the peripheral blood at day 8 was associated with adverse prognosis, with increasing MRD levels being associated with a progressively poorer outcome.[159]
- In multivariate analysis, end of induction therapy MRD was the most powerful prognostic factor, but day 8 peripheral blood MRD maintained its prognostic significance, as did NCI risk group and the presence of favorable trisomies. A smaller study assessed the prognostic significance of peripheral blood MRD at day 15 after 1 week of a steroid prophase and 1 week of multiagent induction therapy.[251] This study also observed multivariate significance for peripheral blood MRD levels after 1 week of multiagent induction therapy.
Both studies identified a group of patients who achieved low MRD levels after 1 week of multiagent induction therapy who had a low rate of subsequent treatment failure. Marrow morphology at the end of induction (induction failure) The vast majority of children with ALL achieve complete morphologic remission by the end of the first month of treatment. The presence of greater than 5% lymphoblasts at the end of the induction phase is observed in 1% to 2% of children with ALL.[21,22,252,253] Patients at highest risk of induction failure have one or more of the following features:[254,255] - T-cell phenotype (especially without a mediastinal mass).
- Precursor B-cell ALL with very high presenting leukocyte counts.
- MLL (KMT2A) gene rearrangement.
- Older age.
- Philadelphia chromosome (before the use of tyrosine kinase inhibitors).
In a large retrospective study, the OS of patients with induction failure was only 32%.[252] However, there was significant clinical and biological heterogeneity. A relatively favorable outcome was observed in patients with precursor B-cell ALL between the ages of 1 and 5 years without adverse cytogenetics (MLL [KMT2A] rearrangement or BCR-ABL1). This group had a 10-year survival exceeding 50%, and HSCT in first remission was not associated with a survival advantage compared with chemotherapy alone for this subset. Patients with the poorest outcomes (<20% 10-year survival) included those who were aged 14 to 18 years, or who had the Philadelphia chromosome or MLL rearrangement. B-cell ALL patients younger than 6 years and T-cell ALL patients (regardless of age) appeared to have better outcomes if treated with allogeneic HSCT after achieving CR than those who received further treatment with chemotherapy alone. Some investigators have suggested that the definition of induction failure should be expanded to include end-of-induction MRD of more than 5%, regardless of morphologic findings. In the UKALL2003 (NCT00222612) study, 59 of 3,113 patients (1.9%) had morphologic induction failure; the 5-year EFS was 51%, and the OS was 58%. However, 2.3% of patients had a morphologic remission, with MRD of 5% or more measured by real-time quantitative IgH-T-cell receptor (TCR) PCR; this group had a 5-year EFS of 47%, similar to those with morphologic induction failure. The authors suggest that using both morphologic and MRD criteria to define induction failure more precisely identifies patients with poor outcomes.[256] Prognostic (Risk) Groups For decades, clinical trial groups studying childhood ALL have utilized risk classification schemes to assign patients to therapeutic regimens based on their estimated risk of treatment failure. Initial risk classification systems utilized clinical factors such as age and presenting WBC count. Response to therapy measures were subsequently added, with some groups utilizing early morphologic bone marrow response (e.g., at day 8 or day 15) and with other groups utilizing response of circulating leukemia cells to single agent prednisone. Modern risk classification systems continue to utilize clinical factors such as age and presenting WBC count, and in addition, incorporate cytogenetics and genomic lesions of leukemia cells at diagnosis (e.g., favorable and unfavorable translocations) and response to therapy based on detection of MRD at end of induction (and in some cases at later time points).[243] The risk classification systems of the COG and the BFM groups are briefly described below. Children's Oncology Group (COG) risk groups In COG protocols, children with ALL are initially stratified into treatment groups (with varying degrees of risk of treatment failure) based on a subset of prognostic factors, including the following: - Age.
- WBC count at diagnosis.
- Immunophenotype.
- Cytogenetics/genomic alterations.
- Presence of extramedullary disease.
- Down syndrome.
- Steroid pretreatment.
EFS rates exceed 85% in children meeting good-risk criteria (aged 1 to <10 years, WBC count <50,000/μL, and precursor B-cell immunophenotype); in children meeting high-risk criteria, EFS rates are approximately 75%.[4,50,249,257,258] Additional factors, including cytogenetic abnormalities and measures of early response to therapy (e.g., day 7 and/or day 14 marrow blast percentage for patients with Down syndrome and MRD levels in peripheral blood on day 8 and in bone marrow samples at the end of induction), considered in conjunction with presenting age, WBC count, immunophenotype, the presence of extramedullary disease, and steroid pretreatment can identify patient groups for postinduction therapy with expected EFS rates ranging from less than 40% to more than 95%.[4,159] Patients who are at very high risk of treatment failure include the following: [259,260,261,262] - Infants with MLL (KMT2A) rearrangements.
- Patients with hypodiploidy (<44 chromosomes).
- Patients with initial induction failure.
Berlin-Frankfurt-Münster (BFM) risk groups Since 2000, risk stratification on BFM protocols has been based almost solely on treatment response criteria. In addition to prednisone prophase response, treatment response is assessed via MRD measurements at two time points, end induction (week 5) and end consolidation (week 12). The BFM risk groups include the following:[240] - Standard risk: Patients who are MRD-negative (i.e., <10-4) at both time points are classified as standard risk.
- Intermediate risk: Patients who have positive MRD at week 5 and low MRD (<10-3) at week 12 are considered intermediate risk.
- High risk: Patients with high MRD (≥10-3) at week 12 are high risk. Patients with a poor response to the prednisone prophase are also considered high risk, regardless of subsequent MRD.
Phenotype, leukemic cell mass estimate, also known as BFM risk factor, and CNS status at diagnosis do not factor into the current risk classification schema. However, patients with either the t(9;22)(q34;q11.2) or the t(4;11)(q21;q23) are considered high risk, regardless of early response measures. Prognostic (risk) groups under clinical evaluation COG AALL08B1(Classification of Newly Diagnosed ALL): COG protocol AALL08B1 stratifies four risk groups for patients with precursor B-cell ALL (low risk, average risk, high risk, and very high risk) based on the following criteria:[1] - Age and presenting leukocyte count (using NCI risk-group criteria).[3]
- Extramedullary disease (presence or absence of CNS and/or testicular leukemia).
- Genomic alterations in leukemia cells.
- Day 8 peripheral blood MRD.
- Day 29 bone marrow morphologic response and MRD.
- Down syndrome.
- Steroid pretreatment.
Morphologic assessment of early response in the bone marrow is no longer performed on days 8 and 15 of induction as part of risk stratification. Patients with T-cell phenotype are treated on a separate study and are not risk classified in this way. For patients with precursor B-cell ALL: - Favorable genetics are defined as the presence of either hyperdiploidy with trisomies of chromosomes 4 and 10 (double trisomy) or the ETV6-RUNX1 fusion.
- Unfavorable characteristics are defined as CNS3 status at diagnosis, induction failure (M3 marrow at day 29), age 13 years and older, and the following unfavorable genomic alterations: hypodiploidy (<44 chromosomes or DNA index <0.81), MLL (KMT2A) rearrangement, t(17;19), and iAMP21. The presence of any of these unfavorable characteristics is sufficient to classify a patient as very high risk, regardless of other presenting features. Infants and children with BCR-ABL1 (Ph+ ALL) are treated on a separate clinical trial.
- MRD levels at day 8 from peripheral blood and at day 29 from bone marrow are used in risk classification.
The four risk groups for precursor B-cell ALL are defined in Table 1.[1] Table 1. Risk Groups for Precursor B-cell Acute Lymphoblastic Leukemia | Low Risk | Average Risk | High Risk | Very High Risk |
|---|
| EFS = event-free survival; HR = age and WBC count risk group is high risk; MRD = minimal residual disease; NCI = National Cancer Institute; PB = peripheral blood; SR = age/WBC count risk group is standard risk; WBC = white blood cell. | | NCI Risk (Age/WBC) | SR | SR | SR | SR | SR | HR (age <13 y) | SR | HR | HR (age ≥13 y) | SR or HR | | Favorable Genetics | Yes | Yes | No | Yes | No | Any | No | Any | Any | Any | | Unfavorable Characteristics | None | None | None | None | None | None | None | None | None | Yes | | Day 8 PB MRD | <0.01% | ≥0.01% | <1% | Any Level | ≥1% | Any Level | Any Level | Any Level | Any Level | Any Level | | Day 29 Marrow MRD | <0.01% | <0.01% | <0.01% | ≥0.01% | <0.01% | <0.01% | ≥0.01% | ≥0.01% | <0.01% | Any Level | | % of Patients (Estimated) | 15% | 36% | 25% | 24% | | Anticipated 5-year EFS | >95% | 90%-95% | 88%-90% | <80% | NCI-2014-00712; AALL1231 (NCT02112916)(Combination Chemotherapy With or Without Bortezomib in Treating Younger Patients With Newly Diagnosed T-Cell ALL or Stage II-IV T-Cell Lymphoblastic Lymphoma): For patients with T-cell ALL, COG uses the following criteria to assign risk category: Standard risk
- M1 marrow with MRD <0.01% on day 29.
- CNS1 status and no testicular disease at diagnosis.
- No steroid therapy pretreatment.
Intermediate risk
- M1 or M2 marrow at day 29 with MRD ≥0.01%.
- MRD <0.1% at end of consolidation.
- Any CNS status at diagnosis.
Very high risk
- M3 marrow at day 29 or MRD ≥0.1% at end of consolidation.
- Any CNS status.
SJCRH (Total XVI): Patients are classified into one of three categories (low, standard, or high risk) based on the presenting age, leukocyte count, presence or absence of CNS3 status or testicular leukemia, immunophenotype, cytogenetics and molecular genetics, DNA index, and early response to therapy. Hence, definitive risk assignment (for provisional low-risk or standard-risk cases based on presenting features) will be made after completion of remission induction therapy. The criteria and the estimated proportion of patients in each category (based on data from the Total XV study) are provided below. Criteria for low-risk ALL (approximately 48% of patients)
- Precursor B-cell ALL with DNA index ≥1.16, ETV6-RUNX1 fusion, or age 1 to 9.9 years and presenting WBC <50 × 109 /L.
- Must not have:
- CNS3 status (≥5 WBC/µl of CSF with morphologically identifiable blasts or cranial nerve palsy).
- Overt testicular leukemia (evidenced by ultrasonogram).
- Adverse genetic features-t(9;22)(q34;q11.2) or BCR-ABL1 fusion; t(1;19) with E2A-PBX1 fusion; rearranged MLL (KMT2A) (as measured by FISH and/or PCR); or hypodiploidy (<44 chromosomes).
- Poor early response (≥1% lymphoblasts on day 15 of remission induction, ≥0.01% lymphoblasts by immunologic or molecular methods on remission date).
Criteria for standard-risk ALL (approximately 44% of patients)
- All cases of T-cell ALL and those of precursor B-cell ALL that do not meet the criteria for low-risk or high-risk ALL.
Criteria for high-risk ALL (approximately 8% of patients)
- t(9;22)(q34;q11.2) or BCR-ABL1 fusion.
- Infants with t(4;11)(q21;q23) or MLL (KMT2A) fusion.
- Induction failure or >1% leukemia lymphoblasts in the bone marrow on remission date.
- >0.1% leukemic lymphoblasts in the bone marrow in week 7 of continuation treatment (i.e., before reinduction 1, about 14 weeks postremission induction).
- Re-emergence of leukemic lymphoblasts by MRD (at any level) in patients previously MRD negative.
- Persistently detectable MRD at lower levels.
- Early T-cell precursor ALL, defined by low expression of T-cell markers together with aberrant expression of myeloid markers.[120] The following features characterize early T-cell precursor ALL:
- Levels of CD5 expression at least tenfold lower than that of normal peripheral blood T-lymphocytes. In the study that identified this subset of T-cell ALL, CD5 expression was tenfold to more than 200-fold lower than that of normal lymphocytes and median percentage of leukemic cells expressing CD5 in the 17 atypical cases was 45%; in contrast to more than 98% for the 122 cases in the typical group.
- Absence (<10%) of CD1a and CD8 expression.
- Expression of cytoplasmic CD3 together with the expression of one or more markers associated with myeloid leukemia such as HLA-Dr, CD34, CD13, CD33, or CD11b, while myeloperoxidase is less than 3% by cytochemistry and/or flow cytometry.
DFCI ALL 16-001 (NCT03020030) (Risk Classification Schemes in Identifying Better Treatment Options for Children and Adolescents with ALL): Patients are assigned an initial risk group by day 10 of therapy on the basis of presenting features and leukemia biology: - Initial low risk: All of the following criteria are met: B-cell ALL, age 1 to younger than 15 years, WBC count less than 50 x 109 /L, CNS1 or CNS2, no intrachromosomal amplification of chromosome 21 (iAMP21), no very high-risk features.
- Initial high risk: Any of the following criteria are met: Aged 15 years or older, WBC count greater than 50 x 109 /L, T-cell ALL, CNS3, presence of iAMP21. Very high-risk features must be absent.
- Initial very high risk: Any of the following criteria are met: IKZF1 deletion, MLL gene-rearrangement, low hypodiploidy (<40 chromosomes).
Patients with BCR-ABL1 are removed from protocol therapy at day 15. The final risk group is based on the initial risk group and MRD (assessed by next-generation sequencing) at the end of induction (day 32; first time point) and week 10 of therapy (second time point): - Final low risk: Initial low risk and MRD less than104 at the first time point.
- Final high risk: Initial low risk with MRD greater than 104 at the first time point and less than 103 at the second time point or initial high risk with MRD less than 103 at the second time point.
- Final very high risk: Initial very high-risk patients or any patient with MRD greater than 103 at the second time point.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with childhood acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Hunger SP, Loh ML, Whitlock JA, et al.: Children's Oncology Group's 2013 blueprint for research: acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (6): 957-63, 2013.
-
Hunger SP, Mullighan CG: Acute Lymphoblastic Leukemia in Children. N Engl J Med 373 (16): 1541-52, 2015.
-
Smith M, Arthur D, Camitta B, et al.: Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol 14 (1): 18-24, 1996.
-
Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.
-
Jeha S, Coustan-Smith E, Pei D, et al.: Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 120 (10): 1514-9, 2014.
-
Vrooman LM, Silverman LB: Childhood acute lymphoblastic leukemia: update on prognostic factors. Curr Opin Pediatr 21 (1): 1-8, 2009.
-
Möricke A, Zimmermann M, Reiter A, et al.: Prognostic impact of age in children and adolescents with acute lymphoblastic leukemia: data from the trials ALL-BFM 86, 90, and 95. Klin Padiatr 217 (6): 310-20, 2005 Nov-Dec.
-
Reaman GH, Sposto R, Sensel MG, et al.: Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children's Cancer Group. J Clin Oncol 17 (2): 445-55, 1999.
-
Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
-
Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.
-
Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.
-
Chessells JM, Harrison CJ, Watson SL, et al.: Treatment of infants with lymphoblastic leukaemia: results of the UK Infant Protocols 1987-1999. Br J Haematol 117 (2): 306-14, 2002.
-
Isoyama K, Eguchi M, Hibi S, et al.: Risk-directed treatment of infant acute lymphoblastic leukaemia based on early assessment of MLL gene status: results of the Japan Infant Leukaemia Study (MLL96). Br J Haematol 118 (4): 999-1010, 2002.
-
Nagayama J, Tomizawa D, Koh K, et al.: Infants with acute lymphoblastic leukemia and a germline MLL gene are highly curable with use of chemotherapy alone: results from the Japan Infant Leukemia Study Group. Blood 107 (12): 4663-5, 2006.
-
Sam TN, Kersey JH, Linabery AM, et al.: MLL gene rearrangements in infant leukemia vary with age at diagnosis and selected demographic factors: a Children's Oncology Group (COG) study. Pediatr Blood Cancer 58 (6): 836-9, 2012.
-
Kang H, Wilson CS, Harvey RC, et al.: Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (8): 1872-81, 2012.
-
Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
-
Stam RW, Schneider P, de Lorenzo P, et al.: Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood 110 (7): 2774-5, 2007.
-
De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
-
Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
-
Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
-
Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
-
Forestier E, Schmiegelow K; on behalf of the Nordic Society of Paediatric Haematology and Oncology NOPHO: The incidence peaks of the childhood acute leukemias reflect specific cytogenetic aberrations. J Pediatr Hematol Oncol 28 (8): 486-95, 2006.
-
Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
-
Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.
-
Pulte D, Gondos A, Brenner H: Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 113 (7): 1408-11, 2009.
-
Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.
-
Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 28. Also available online. Last accessed January 27, 2017.
-
Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013, Section 29. Also available online. Last accessed May 31, 2017.
-
Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34. Also available online. Last accessed January 27, 2017.
-
de Bont JM, Holt B, Dekker AW, et al.: Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia 18 (12): 2032-5, 2004.
-
Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.
-
Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.
-
Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.
-
Pullen J, Shuster JJ, Link M, et al.: Significance of commonly used prognostic factors differs for children with T cell acute lymphocytic leukemia (ALL), as compared to those with B-precursor ALL. A Pediatric Oncology Group (POG) study. Leukemia 13 (11): 1696-707, 1999.
-
Goldberg JM, Silverman LB, Levy DE, et al.: Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol 21 (19): 3616-22, 2003.
-
Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
-
Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
-
Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.
-
Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
-
Vaitkevičienė G, Forestier E, Hellebostad M, et al.: High white blood cell count at diagnosis of childhood acute lymphoblastic leukaemia: biological background and prognostic impact. Results from the NOPHO ALL-92 and ALL-2000 studies. Eur J Haematol 86 (1): 38-46, 2011.
-
Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.
-
Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.
-
Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
-
Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.
-
Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.
-
te Loo DM, Kamps WA, van der Does-van den Berg A, et al.: Prognostic significance of blasts in the cerebrospinal fluid without pleiocytosis or a traumatic lumbar puncture in children with acute lymphoblastic leukemia: experience of the Dutch Childhood Oncology Group. J Clin Oncol 24 (15): 2332-6, 2006.
-
Gilchrist GS, Tubergen DG, Sather HN, et al.: Low numbers of CSF blasts at diagnosis do not predict for the development of CNS leukemia in children with intermediate-risk acute lymphoblastic leukemia: a Childrens Cancer Group report. J Clin Oncol 12 (12): 2594-600, 1994.
-
Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.
-
Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
-
Levinsen M, Taskinen M, Abrahamsson J, et al.: Clinical features and early treatment response of central nervous system involvement in childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 61 (8): 1416-21, 2014.
-
Cherlow JM, Sather H, Steinherz P, et al.: Craniospinal irradiation for acute lymphoblastic leukemia with central nervous system disease at diagnosis: a report from the Children's Cancer Group. Int J Radiat Oncol Biol Phys 36 (1): 19-27, 1996.
-
Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.
-
Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.
-
Bassal M, La MK, Whitlock JA, et al.: Lymphoblast biology and outcome among children with Down syndrome and ALL treated on CCG-1952. Pediatr Blood Cancer 44 (1): 21-8, 2005.
-
Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
-
Whitlock JA, Sather HN, Gaynon P, et al.: Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 106 (13): 4043-9, 2005.
-
Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
-
Lundin C, Forestier E, Klarskov Andersen M, et al.: Clinical and genetic features of pediatric acute lymphoblastic leukemia in Down syndrome in the Nordic countries. J Hematol Oncol 7 (1): 32, 2014.
-
Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
-
Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
-
Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
-
Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
-
Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
-
Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
-
Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
-
Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
-
Pui CH, Boyett JM, Relling MV, et al.: Sex differences in prognosis for children with acute lymphoblastic leukemia. J Clin Oncol 17 (3): 818-24, 1999.
-
Shuster JJ, Wacker P, Pullen J, et al.: Prognostic significance of sex in childhood B-precursor acute lymphoblastic leukemia: a Pediatric Oncology Group Study. J Clin Oncol 16 (8): 2854-63, 1998.
-
Chessells JM, Richards SM, Bailey CC, et al.: Gender and treatment outcome in childhood lymphoblastic leukaemia: report from the MRC UKALL trials. Br J Haematol 89 (2): 364-72, 1995.
-
Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.
-
Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.
-
Bhatia S: Influence of race and socioeconomic status on outcome of children treated for childhood acute lymphoblastic leukemia. Curr Opin Pediatr 16 (1): 9-14, 2004.
-
Kadan-Lottick NS, Ness KK, Bhatia S, et al.: Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA 290 (15): 2008-14, 2003.
-
Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
-
Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.
-
Yang JJ, Cheng C, Devidas M, et al.: Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 43 (3): 237-41, 2011.
-
Xu H, Cheng C, Devidas M, et al.: ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol 30 (7): 751-7, 2012.
-
Aldhafiri FK, McColl JH, Reilly JJ: Prognostic significance of being overweight and obese at diagnosis in children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 36 (3): 234-6, 2014.
-
Baillargeon J, Langevin AM, Lewis M, et al.: Obesity and survival in a cohort of predominantly Hispanic children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 28 (9): 575-8, 2006.
-
Hijiya N, Panetta JC, Zhou Y, et al.: Body mass index does not influence pharmacokinetics or outcome of treatment in children with acute lymphoblastic leukemia. Blood 108 (13): 3997-4002, 2006.
-
Butturini AM, Dorey FJ, Lange BJ, et al.: Obesity and outcome in pediatric acute lymphoblastic leukemia. J Clin Oncol 25 (15): 2063-9, 2007.
-
Gelelete CB, Pereira SH, Azevedo AM, et al.: Overweight as a prognostic factor in children with acute lymphoblastic leukemia. Obesity (Silver Spring) 19 (9): 1908-11, 2011.
-
Orgel E, Sposto R, Malvar J, et al.: Impact on survival and toxicity by duration of weight extremes during treatment for pediatric acute lymphoblastic leukemia: A report from the Children's Oncology Group. J Clin Oncol 32 (13): 1331-7, 2014.
-
Orgel E, Tucci J, Alhushki W, et al.: Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood 124 (26): 3932-8, 2014.
-
den Hoed MA, Pluijm SM, de Groot-Kruseman HA, et al.: The negative impact of being underweight and weight loss on survival of children with acute lymphoblastic leukemia. Haematologica 100 (1): 62-9, 2015.
-
Bennett JM, Catovsky D, Daniel MT, et al.: The morphological classification of acute lymphoblastic leukaemia: concordance among observers and clinical correlations. Br J Haematol 47 (4): 553-61, 1981.
-
Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008.
-
Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
-
Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
-
Möricke A, Ratei R, Ludwig WD, et al.: Prognostic factors in CD10 negative precursor b-cell acute lymphoblastic leukemia in children: data from three consecutive trials ALL-BFM 86, 90, and 95. [Abstract] Blood 104 (11): A-1957, 540a, 2004.
-
Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
-
Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
-
Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.
-
Slack JL, Arthur DC, Lawrence D, et al.: Secondary cytogenetic changes in acute promyelocytic leukemia--prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol 15 (5): 1786-95, 1997.
-
Attarbaschi A, Mann G, Dworzak M, et al.: Mediastinal mass in childhood T-cell acute lymphoblastic leukemia: significance and therapy response. Med Pediatr Oncol 39 (6): 558-65, 2002.
-
Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.
-
Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.
-
Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.
-
van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.
-
Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.
-
Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.
-
Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.
-
Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.
-
Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.
-
Breit S, Stanulla M, Flohr T, et al.: Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108 (4): 1151-7, 2006.
-
Kox C, Zimmermann M, Stanulla M, et al.: The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia 24 (12): 2005-13, 2010.
-
Jenkinson S, Koo K, Mansour MR, et al.: Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia 27 (1): 41-7, 2013.
-
Fogelstrand L, Staffas A, Wasslavik C, et al.: Prognostic implications of mutations in NOTCH1 and FBXW7 in childhood T-ALL treated according to the NOPHO ALL-1992 and ALL-2000 protocols. Pediatr Blood Cancer 61 (3): 424-30, 2014.
-
Larson Gedman A, Chen Q, Kugel Desmoulin S, et al.: The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. Leukemia 23 (8): 1417-25, 2009.
-
Zuurbier L, Homminga I, Calvert V, et al.: NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia 24 (12): 2014-22, 2010.
-
Clappier E, Collette S, Grardel N, et al.: NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia 24 (12): 2023-31, 2010.
-
Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.
-
Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.
-
Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.
-
Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.
-
Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.
-
Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.
-
Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.
-
Coustan-Smith E, Mullighan CG, Onciu M, et al.: Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol 10 (2): 147-56, 2009.
-
Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.
-
Ma M, Wang X, Tang J, et al.: Early T-cell precursor leukemia: a subtype of high risk childhood acute lymphoblastic leukemia. Front Med 6 (4): 416-20, 2012.
-
Inukai T, Kiyokawa N, Campana D, et al.: Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children's Cancer Study Group Study L99-15. Br J Haematol 156 (3): 358-65, 2012.
-
Patrick K, Wade R, Goulden N, et al.: Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br J Haematol 166 (3): 421-4, 2014.
-
Wood BL, Winter SS, Dunsmore KP, et al.: T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children's Oncology Group (COG) study AALL0434. [Abstract] Blood 124 (21): A-1, 2014. Also available online. Last accessed January 27, 2017.
-
Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.
-
Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.
-
Pui CH, Rubnitz JE, Hancock ML, et al.: Reappraisal of the clinical and biologic significance of myeloid-associated antigen expression in childhood acute lymphoblastic leukemia. J Clin Oncol 16 (12): 3768-73, 1998.
-
Uckun FM, Sather HN, Gaynon PS, et al.: Clinical features and treatment outcome of children with myeloid antigen positive acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 90 (1): 28-35, 1997.
-
Gerr H, Zimmermann M, Schrappe M, et al.: Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol 149 (1): 84-92, 2010.
-
Rubnitz JE, Onciu M, Pounds S, et al.: Acute mixed lineage leukemia in children: the experience of St Jude Children's Research Hospital. Blood 113 (21): 5083-9, 2009.
-
Al-Seraihy AS, Owaidah TM, Ayas M, et al.: Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica 94 (12): 1682-90, 2009.
-
Bene MC, Castoldi G, Knapp W, et al.: Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 9 (10): 1783-6, 1995.
-
Vardiman JW, Thiele J, Arber DA, et al.: The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114 (5): 937-51, 2009.
-
Borowitz MJ, Béné MC, Harris NL: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008, pp 150-5.
-
Matutes E, Pickl WF, Van't Veer M, et al.: Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood 117 (11): 3163-71, 2011.
-
Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.
-
Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.
-
Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.
-
Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.
-
Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.
-
Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.
-
Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.
-
Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.
-
Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.
-
Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.
-
Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.
-
Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.
-
Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.
-
Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.
-
Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.
-
Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.
-
Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
-
Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
-
Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.
-
Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.
-
Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.
-
Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.
-
Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
-
Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.
-
Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.
-
Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.
-
Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.
-
Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.
-
Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.
-
Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
-
Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.
-
van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.
-
Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
-
Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
-
Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.
-
Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
-
Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
-
Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
-
Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.
-
Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.
-
Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.
-
Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.
-
Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.
-
Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.
-
Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.
-
Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.
-
Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.
-
Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.
-
Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.
-
Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.
-
Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.
-
Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.
-
Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.
-
Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.
-
Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.
-
Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.
-
Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.
-
Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.
-
Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.
-
Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.
-
Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.
-
Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
-
Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.
-
Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.
-
Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
-
Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.
-
Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.
-
Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.
-
Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
-
Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.
-
Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.
-
van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.
-
Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.
-
Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.
-
van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.
-
Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.
-
Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.
-
Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.
-
Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
-
Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.
-
Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.
-
Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.
-
Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.
-
Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.
-
Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.
-
Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.
-
Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.
-
Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.
-
Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.
-
Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.
-
Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.
-
Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
-
Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
-
Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
-
Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.
-
Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.
-
Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.
-
Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.
-
Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.
-
Relling MV, Dervieux T: Pharmacogenetics and cancer therapy. Nat Rev Cancer 1 (2): 99-108, 2001.
-
van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.
-
Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.
-
Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.
-
Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.
-
Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.
-
Pui CH, Pei D, Coustan-Smith E, et al.: Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol 16 (4): 465-74, 2015.
-
Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.
-
Bartram J, Wade R, Vora A, et al.: Excellent outcome of minimal residual disease-defined low-risk patients is sustained with more than 10 years follow-up: results of UK paediatric acute lymphoblastic leukaemia trials 1997-2003. Arch Dis Child 101 (5): 449-54, 2016.
-
Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.
-
Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
-
Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.
-
Borowitz MJ, Wood BL, Devidas M, et al.: Assessment of end induction minimal residual disease (MRD) in childhood B precursor acute lymphoblastic leukemia (ALL) to eliminate the need for day 14 marrow examination: A Children's Oncology Group study. [Abstract] J Clin Oncol 31 (Suppl 15): A-10001, 2013. Also available online. Last accessed January 27, 2017.
-
Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
-
Griffin TC, Shuster JJ, Buchanan GR, et al.: Slow disappearance of peripheral blood blasts is an adverse prognostic factor in childhood T cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Leukemia 14 (5): 792-5, 2000.
-
Volejnikova J, Mejstrikova E, Valova T, et al.: Minimal residual disease in peripheral blood at day 15 identifies a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with superior prognosis. Haematologica 96 (12): 1815-21, 2011.
-
Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.
-
Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
-
Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.
-
Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.
-
O'Connor D, Moorman AV, Wade R, et al.: Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol 35 (6): 660-667, 2017.
-
Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
-
Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
-
Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.
-
Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.
-
Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.
-
Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.
Treatment Option Overview for Childhood ALLSpecial Considerations for the Treatment of Children with Cancer Because treatment of children with ALL entails complicated risk assignment and therapies and the need for intensive supportive care (e.g., transfusions; management of infectious complications; and emotional, financial, and developmental support), evaluation and treatment are best coordinated by a multidisciplinary team in cancer centers or hospitals with all of the necessary pediatric supportive care facilities. [1] A multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life: - Primary care physicians.
- Pediatric surgical subspecialists.
- Radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child life professionals.
- Psychologists.
Guidelines for cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[1] Treatment of childhood ALL typically involves chemotherapy given for 2 to 3 years. Because myelosuppression and generalized immunosuppression are anticipated consequences of leukemia and chemotherapy treatment, adequate facilities must be immediately available both for hematologic support and for the treatment of infections and other complications throughout all phases of therapy. Approximately 1% to 3% of patients die during induction therapy and another 1% to 3% die during the initial remission from treatment-related complications.[2,3,4,5] It is important that the clinical centers and the specialists directing the patient's care maintain contact with the referring physician in the community. Strong lines of communication optimize any urgent or interim care required when the child is at home. Clinical trials are generally available for children with ALL, with specific protocols designed for children at standard (low) risk of treatment failure and for children at higher risk of treatment failure. Clinical trials for children with ALL are generally designed to compare therapy that is currently accepted as standard for a particular risk group with a potentially better treatment approach that may improve survival outcome and/or diminish toxicities associated with the standard treatment regimen. Many of the therapeutic innovations that produced increased survival rates in children with ALL were established through clinical trials, and it is appropriate for children and adolescents with ALL to be offered participation in a clinical trial. Risk-based treatment assignment is an important therapeutic strategy utilized for children with ALL. This approach allows children who historically have a very good outcome to be treated with less intensive therapy and to be spared more toxic treatments, while allowing children with a historically lower probability of long-term survival to receive more intensive therapy that may increase their chance of cure. (Refer to the Risk-Based Treatment Assignment section of this summary for more information about a number of clinical and laboratory features that have demonstrated prognostic value.) Phases of Therapy Treatment for children with ALL is typically divided as follows: - Remission induction (at the time of diagnosis).
- Postinduction therapy (after achieving complete remission).
- Consolidation/intensification therapy.
- Maintenance or continuation therapy.
Sanctuary Sites Historically, certain extramedullary sites have been considered sanctuary sites (i.e., anatomic spaces that are poorly penetrated by many of the orally and intravenously administered chemotherapy agents typically used to treat ALL). The two most important sanctuary sites in childhood ALL are the central nervous system (CNS) and the testes. Successful treatment of ALL requires therapy that effectively addresses clinical or subclinical involvement of leukemia in these extramedullary sanctuary sites. Central nervous system (CNS) At diagnosis, approximately 3% of patients have CNS3 disease (defined as cerebrospinal fluid specimen with ≥5 white blood cells/μL with lymphoblasts and/or the presence of cranial nerve palsies). However, unless specific therapy is directed toward the CNS, most children will eventually develop overt CNS leukemia whether or not lymphoblasts were detected in the spinal fluid at initial diagnosis. CNS-directed treatments include intrathecal chemotherapy, CNS-directed systemic chemotherapy, and cranial radiation; some or all of these are included in current regimens for ALL. (Refer to the CNS-Directed Therapy for Childhood ALL section of this summary for more information.) Testes Overt testicular involvement at the time of diagnosis occurs in approximately 2% of males. In early ALL trials, testicular involvement at diagnosis was an adverse prognostic factor. With more aggressive initial therapy, however, the prognostic significance of initial testicular involvement is unclear.[6,7] The role of radiation therapy for testicular involvement is also unclear. A study from St. Jude Children's Research Hospital suggests that a good outcome can be achieved with aggressive conventional chemotherapy without radiation.[6] The Children's Oncology Group has also adopted this strategy for boys with testicular involvement that resolves completely during induction chemotherapy. References:
-
Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.
-
Rubnitz JE, Lensing S, Zhou Y, et al.: Death during induction therapy and first remission of acute leukemia in childhood: the St. Jude experience. Cancer 101 (7): 1677-84, 2004.
-
Christensen MS, Heyman M, Möttönen M, et al.: Treatment-related death in childhood acute lymphoblastic leukaemia in the Nordic countries: 1992-2001. Br J Haematol 131 (1): 50-8, 2005.
-
Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
-
Lund B, Åsberg A, Heyman M, et al.: Risk factors for treatment related mortality in childhood acute lymphoblastic leukaemia. Pediatr Blood Cancer 56 (4): 551-9, 2011.
-
Hijiya N, Liu W, Sandlund JT, et al.: Overt testicular disease at diagnosis of childhood acute lymphoblastic leukemia: lack of therapeutic role of local irradiation. Leukemia 19 (8): 1399-403, 2005.
-
Sirvent N, Suciu S, Bertrand Y, et al.: Overt testicular disease (OTD) at diagnosis is not associated with a poor prognosis in childhood acute lymphoblastic leukemia: results of the EORTC CLG Study 58881. Pediatr Blood Cancer 49 (3): 344-8, 2007.
Treatment for Newly Diagnosed Childhood ALLStandard Treatment Options for Newly Diagnosed ALL Standard treatment options for newly diagnosed childhood acute lymphoblastic leukemia (ALL) include the following: - Chemotherapy.
Remission induction chemotherapy The goal of the first phase of therapy (remission induction) is to induce a complete remission (CR). This phase typically lasts 4 weeks. Overall, approximately 98% of patients with newly diagnosed precursor B-cell ALL achieve CR by the end of this phase, with somewhat lower rates in infants and in noninfant patients with T-cell ALL or high presenting leukocyte counts.[1,2,3,4,5] Induction chemotherapy typically consists of the following drugs, with or without an anthracycline (either doxorubicin or daunorubicin): - Vincristine.
- Corticosteroid (either prednisone or dexamethasone).
- L-asparaginase.
The Children's Oncology Group (COG) protocols administer a three-drug induction (vincristine, corticosteroid, and pegaspargase) to National Cancer Institute (NCI) standard-risk B-cell ALL patients and a four-drug induction (vincristine, corticosteroid, and pegaspargase plus anthracycline) to NCI high-risk B-cell ALL and all T-cell ALL patients. Other groups use a four-drug induction for all patients.[1,2,3] Corticosteroid therapy Many current regimens utilize dexamethasone instead of prednisone during remission induction and later phases of therapy, although controversy exists as to whether dexamethasone benefits all subsets of patients. Some trials also suggest that dexamethasone during induction may be associated with more toxicity than prednisone, including higher rates of infection, myopathy, and behavioral changes.[1,6,7,8] The COG reported that dexamethasone during induction was associated with a higher risk of osteonecrosis in older children (aged >10 years),[8] although this finding has not been confirmed in other randomized studies.[1,7] Evidence (dexamethasone vs. prednisone during induction): - The Children's Cancer Group conducted a randomized trial that compared dexamethasone and prednisone in standard-risk B-cell ALL patients receiving a three-drug induction without anthracycline.[6]
- Dexamethasone was associated with a superior event-free survival (EFS).
- Dexamethasone was associated with a higher frequency of reversible steroid myopathy and hyperglycemia. No significant differences in rates of infection during induction were observed between the two randomized arms.
- Another randomized trial that included both standard-risk and high-risk patients was conducted by the United Kingdom Medical Research Council.[7]
- The trial demonstrated that dexamethasone was associated with a more favorable outcome than prednisolone in all patient subgroups.
- Patients who received dexamethasone had a significantly lower incidence of both central nervous system (CNS) and non-CNS relapses than did patients who received prednisolone.
- Dexamethasone was associated with a higher incidence of steroid-associated behavioral problems and myopathy, but an excess risk of osteonecrosis was not observed. There was no difference in induction death rates between the randomized groups.
- The AIEOP-BFM 2000 trial randomly assigned 3,720 patients to receive either dexamethasone (10 mg/m2 /d) or prednisone (60 mg/m2 /d) during multiagent remission induction (including anthracycline for all patients) after a 7-day prednisone prophase.[9]
- Dexamethasone was associated with higher incidence of life-threatening events (primarily infections), resulting in a significantly higher induction death rate (2.5% for dexamethasone vs. 0.9% for prednisone; P = .00013).
- There was no difference in rates of osteonecrosis between the randomized groups.
- The 5-year cumulative incidence of relapse was significantly lower with dexamethasone (11% vs. 16%; P < .0001), resulting in superior 5-year EFS (84% for dexamethasone vs. 81% for prednisone, P = .024) despite the increased induction death rate.
- No difference in overall survival (OS) was observed based on steroid randomization, although the study was not sufficiently powered to detect small differences in OS.
- The COG conducted a randomized trial of dexamethasone and prednisone in NCI high-risk B-cell ALL patients.[8] Patients were randomly assigned to receive 14 days of dexamethasone or 28 days of prednisone during a four-drug induction (with anthracycline). This trial also included a randomized comparison of high-dose and escalating-dose methotrexate during the interim maintenance phase.
- Dexamethasone was associated with a higher rate of infection, but there was no difference in the induction death rate when comparing dexamethasone and prednisone.
- For patients who were younger than 10 years at diagnosis, there was a significant interaction between the corticosteroid and methotrexate randomizations; however, the best outcome for this group of patients was observed in those who received both dexamethasone during induction and high-dose methotrexate during interim maintenance.
- The corticosteroid randomization was closed early for patients aged 10 years or older at diagnosis because of excessive rates of osteonecrosis in patients randomly assigned to dexamethasone; however, it did not appear that there was any EFS benefit associated with dexamethasone in these older patients (5-year EFS of 73.1% with dexamethasone and 73.9% with prednisone; P = .78)
The ratio of dexamethasone to prednisone dose used may influence outcome. Studies in which the dexamethasone to prednisone ratio was 1:5 to 1:7 have shown a better result for dexamethasone, while studies that used a 1:10 ratio have shown similar outcomes.[10] L-asparaginase Several forms of L-asparaginase have been used in the treatment of children with ALL, including the following: - Pegaspargase (PEG-asparaginase).
- Asparaginase Erwinia chrysanthemi (Erwinia L-asparaginase).
- Native Escherichia coli (E. coli) L-asparaginase.
Only pegaspargase and Erwinia L-asparaginase are available in the United States. Native E. coli L-asparaginase remains available in other countries. Pegaspargase (PEG-asparaginase) Pegaspargase, a form of L-asparaginase in which the E. coli-derived enzyme is modified by the covalent attachment of polyethylene glycol, is the most common preparation used during both induction and postinduction phases of treatment in newly diagnosed patients treated in the United States and Western Europe. Pegaspargase may be given either intramuscularly (IM) or intravenously (IV).[11] Pharmacokinetics and toxicity profiles are similar for IM and IV pegaspargase administration.[11] There is no evidence that IV administration of pegaspargase is more toxic than IM administration.[11,12,13] Pegaspargase has a much longer serum half-life than native E. coli L-asparaginase, producing prolonged asparagine depletion after a single injection.[14] Serum asparaginase enzyme activity levels of more than 0.1 IU/mL have been associated with serum asparagine depletion. Studies have shown that a single dose of pegaspargase given either IM or IV as part of multiagent induction results in serum enzyme activity of more than 0.1 IU/mL in nearly all patients for at least 2 to 3 weeks.[11,12,15] Evidence (use of pegaspargase instead of native E. coli L-asparaginase): - A randomized comparison of IV pegaspargase versus IM native E. coli asparaginase was conducted. Each agent was administered for a 30-week period after the achievement of CR.[13][Level of evidence: 1iiC]
- Serum asparaginase activity (SAA) levels were significantly higher with IV pegaspargase and exceeded goal therapeutic levels (>0.1 IU/mL) in nearly all patients throughout the 30-week period.
- There was no significant difference in EFS and OS between the randomized arms.
- There was no difference in rates of asparaginase-related toxicities, including hypersensitivity, pancreatitis, and thromboembolic complications.
- Similar outcome and similar rates of asparaginase-related toxicities were observed for both groups of patients.
- IV pegaspargase was associated with less treatment-related anxiety, as assessed by patient and parent surveys.
- Another randomized trial of patients with standard-risk ALL assigned patients to receive either pegaspargase or native E. coli asparaginase in induction and each of two delayed intensification courses.[15]
- A single dose of pegaspargase given in conjunction with vincristine and prednisone during induction therapy appeared to have similar activity and toxicity as nine doses of IM E. coli L-asparaginase (3 times a week for 3 weeks).[15]
- The use of pegaspargase was associated with more rapid blast clearance and a lower incidence of neutralizing antibodies.
Patients with an allergic reaction to pegaspargase are typically switched to Erwinia L-asparaginase. Measurement of SAA levels after a mild or questionable reaction to pegaspargase may help to differentiate patients for whom the switch to Erwinia is indicated (because of inadequate SAA) versus those for whom a change in preparation may not be necessary.[16,17] Several studies have identified a subset of patients who experience silent inactivation of asparaginase, defined as absence of therapeutic SAA levels without overt allergy.[18,19] In a trial conducted by the Dana-Farber Cancer Institute (DFCI) Consortium, 12% of patients treated initially with native E.coli L-asparaginase demonstrated silent inactivation; these patients had a superior EFS if their asparaginase preparation was changed.[19] The frequency of silent inactivation in patients initially treated with pegaspargase appears to be low (<10%).[13,18] Determination of the optimal frequency of pharmacokinetic monitoring for pegaspargase-treated patients, and whether such screening impacts outcome, awaits further investigation. AsparaginaseErwinia chrysanthemi(ErwiniaL-asparaginase) Erwinia L-asparaginase is typically used in patients who have experienced allergy to native E. coli or pegaspargase. The half-life of Erwinia L-asparaginase (0.65 days) is much shorter than that of native E. coli (1.2 days) or pegaspargase (5.7 days).[14] If Erwinia L-asparaginase is utilized, the shorter half-life of the Erwinia preparation requires more frequent administration to achieve adequate asparagine depletion. Evidence (increased dose frequency of Erwinia L-asparaginase needed to achieve goal therapeutic effect): - A COG trial demonstrated that IM Erwinia L-asparaginase given three times a week to patients with an allergy to pegaspargase leads to therapeutic serum asparaginase enzyme activity levels (defined as a level ≥0.1 IU/mL). On that trial, 96% of children achieved a level of 0.1 IU/mL or more at 2 days after a dose of Erwinia L-asparaginase and 85% did so at 3 days after a dose.[20]
- A trial of IV Erwinia L-asparaginase given on a Monday-Wednesday-Friday schedule to patients with an allergy to pegaspargase demonstrated therapeutic serum asparaginase enzyme activity (defined as ≥0.1 IU/mL) in 83% of patients 48 hours after a dose but in only 43% of patients 72 hours after a dose. If IV Erwinia is given on a Monday-Wednesday-Friday schedule, the authors suggest that 72-hour nadir enzyme activity levels be monitored to ensure therapeutic levels.[21]
Anthracycline during induction The COG protocols administer a three-drug induction (vincristine, corticosteroid, and pegaspargase) to NCI standard-risk B-cell ALL patients and a four-drug induction (vincristine, corticosteroid, and pegaspargase plus anthracycline) to NCI high-risk B-cell ALL and all T-cell ALL patients. Other groups use a four-drug induction for all patients.[1,2,3] In induction regimens that include an anthracycline, either daunorubicin or doxorubicin are typically utilized. In a randomized trial comparing the two agents during induction, there were no differences in early response measures, including reduction in peripheral blood blast counts during the first week of therapy, day 15 marrow morphology, and end-induction minimal residual disease (MRD) levels.[22][Level of evidence: 1iiDiv] Response to remission induction chemotherapy More than 95% of children with newly diagnosed ALL will achieve a CR within the first 4 weeks of treatment. Of those who fail to achieve CR within the first 4 weeks, approximately one-half will experience a toxic death during the induction phase (usually caused by infection) and the other half will have resistant disease (persistent morphologic leukemia).[23,24,25]; [26][Level of evidence: 3iA] Most patients with persistent leukemia at the end of the 4-week induction phase have a poor prognosis and may benefit from an allogeneic hematopoietic stem cell transplant (HSCT) once CR is achieved.[27,28,4] In a large retrospective series, the 10-year OS for patients with persistent leukemia was 32%.[29] A trend for superior outcome with allogeneic HSCT compared with chemotherapy alone was observed in patients with T-cell phenotype (any age) and precursor B-cell patients younger than 6 years. Precursor B-cell ALL patients who were aged 1 to 5 years at diagnosis and did not have any adverse cytogenetic abnormalities (MLL (KMT2A) rearrangement, BCR-ABL1) had a relatively favorable prognosis, without any advantage in outcome with the utilization of HSCT compared with chemotherapy alone.[29] For patients who achieve CR, measures of the rapidity of blast clearance and MRD determinations have important prognostic significance, particularly the following: - The percentage of morphologically detectable marrow blasts at 7 and 14 days after starting multiagent remission induction therapy has been correlated with relapse risk,[30] and has been used in the past by the COG to risk-stratify patients. However, in multivariate analyses, when end-induction MRD is included, these early marrow findings lose their prognostic significance.[31,32]
- End-induction levels of submicroscopic MRD, assessed either by multiparameter flow cytometry or polymerase chain reaction, strongly correlates with long-term outcome.[31,33,34,35] Intensification of postinduction therapy for patients with high levels of end-induction MRD is a common component of most ALL treatment regimens. In a randomized trial conducted by the UK-ALL group, augmented postinduction therapy was shown to improve outcome for standard-risk and intermediate-risk patients with high end-induction MRD.[36]
- MRD levels earlier in induction (e.g., days 8 and 15) and at later postinduction time points (e.g., week 12 after starting therapy) have also been shown to have prognostic significance in both B-cell and T-cell ALL.[31,32,35,37,38,39,40]
(Refer to the Response to initial treatment section of this summary for more information.) (Refer to the CNS-Directed Therapy for Childhood ALL section of this summary for specific information about CNS therapy to prevent CNS relapse in children with newly diagnosed ALL.) Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with untreated childhood acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
-
Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
-
Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
-
Oudot C, Auclerc MF, Levy V, et al.: Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: the FRALLE 93 study. J Clin Oncol 26 (9): 1496-503, 2008.
-
Salzer WL, Devidas M, Carroll WL, et al.: Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984-2001: a report from the children's oncology group. Leukemia 24 (2): 355-70, 2010.
-
Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.
-
Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.
-
Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.
-
Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
-
McNeer JL, Nachman JB: The optimal use of steroids in paediatric acute lymphoblastic leukaemia: no easy answers. Br J Haematol 149 (5): 638-52, 2010.
-
Silverman LB, Supko JG, Stevenson KE, et al.: Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood 115 (7): 1351-3, 2010.
-
Rizzari C, Citterio M, Zucchetti M, et al.: A pharmacological study on pegylated asparaginase used in front-line treatment of children with acute lymphoblastic leukemia. Haematologica 91 (1): 24-31, 2006.
-
Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
-
Asselin BL, Whitin JC, Coppola DJ, et al.: Comparative pharmacokinetic studies of three asparaginase preparations. J Clin Oncol 11 (9): 1780-6, 1993.
-
Avramis VI, Sencer S, Periclou AP, et al.: A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood 99 (6): 1986-94, 2002.
-
van der Sluis IM, Vrooman LM, Pieters R, et al.: Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica 101 (3): 279-85, 2016.
-
Bleyer A, Asselin BL, Koontz SE, et al.: Clinical application of asparaginase activity levels following treatment with pegaspargase. Pediatr Blood Cancer 62 (6): 1102-5, 2015.
-
Tong WH, Pieters R, Kaspers GJ, et al.: A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood 123 (13): 2026-33, 2014.
-
Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
-
Salzer WL, Asselin B, Supko JG, et al.: Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood 122 (4): 507-14, 2013.
-
Vrooman LM, Kirov II, Dreyer ZE, et al.: Activity and Toxicity of Intravenous Erwinia Asparaginase Following Allergy to E. coli-Derived Asparaginase in Children and Adolescents With Acute Lymphoblastic Leukemia. Pediatr Blood Cancer 63 (2): 228-33, 2016.
-
Escherich G, Zimmermann M, Janka-Schaub G, et al.: Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer 60 (2): 254-7, 2013.
-
Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.
-
Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
-
Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
-
Prucker C, Attarbaschi A, Peters C, et al.: Induction death and treatment-related mortality in first remission of children with acute lymphoblastic leukemia: a population-based analysis of the Austrian Berlin-Frankfurt-Münster study group. Leukemia 23 (7): 1264-9, 2009.
-
Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.
-
Silverman LB, Gelber RD, Young ML, et al.: Induction failure in acute lymphoblastic leukemia of childhood. Cancer 85 (6): 1395-404, 1999.
-
Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.
-
Gaynon PS, Desai AA, Bostrom BC, et al.: Early response to therapy and outcome in childhood acute lymphoblastic leukemia: a review. Cancer 80 (9): 1717-26, 1997.
-
Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
-
Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.
-
van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.
-
Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.
-
Conter V, Bartram CR, Valsecchi MG, et al.: Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 115 (16): 3206-14, 2010.
-
Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.
-
Coustan-Smith E, Sancho J, Behm FG, et al.: Prognostic importance of measuring early clearance of leukemic cells by flow cytometry in childhood acute lymphoblastic leukemia. Blood 100 (1): 52-8, 2002.
-
Basso G, Veltroni M, Valsecchi MG, et al.: Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J Clin Oncol 27 (31): 5168-74, 2009.
-
Schrappe M, Valsecchi MG, Bartram CR, et al.: Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood 118 (8): 2077-84, 2011.
-
Karsa M, Dalla Pozza L, Venn NC, et al.: Improving the identification of high risk precursor B acute lymphoblastic leukemia patients with earlier quantification of minimal residual disease. PLoS One 8 (10): e76455, 2013.
Postinduction Treatment for Childhood ALLStandard Postinduction Treatment Options for Childhood ALL Standard treatment options for consolidation/intensification and maintenance therapy include the following: - Chemotherapy.
Central nervous system (CNS)-directed therapy is provided during premaintenance chemotherapy by all groups. Some protocols (Children's Oncology Group [COG], St. Jude Children's Research Hospital [SJCRH], and Dana-Farber Cancer Institute [DFCI]) provide ongoing intrathecal chemotherapy during maintenance, while others (Berlin-Frankfurt-Münster [BFM]) do not. (Refer to the CNS-Directed Therapy for Childhood ALL section of this summary for specific information about CNS therapy to prevent CNS relapse in children with acute lymphoblastic leukemia [ALL] who are receiving postinduction therapy.) Consolidation/intensification therapy Once complete remission (CR) has been achieved, systemic treatment in conjunction with CNS-directed therapy follows. The intensity of the postinduction chemotherapy varies considerably depending on risk group assignment, but all patients receive some form of intensification after the achievement of CR and before beginning maintenance therapy. The most commonly used intensification schema is the BFM backbone. This therapeutic backbone, first introduced by the BFM clinical trials group, includes the following:[1] - An initial consolidation (referred to as Induction IB) immediately after the initial induction phase. This phase includes cyclophosphamide, low-dose cytarabine, and mercaptopurine.
- An interim maintenance phase, which includes multiple doses of high-dose methotrexate (typically 5 g/m2) with leucovorin rescue or escalating doses of methotrexate (starting dose 100 mg/m2) without leucovorin rescue.
- Reinduction (or delayed intensification), which typically includes agents and schedules similar to those used during the induction and initial consolidation phases.
- Maintenance, typically consisting of daily mercaptopurine, weekly low-dose methotrexate, and sometimes, administration of vincristine and a corticosteroid, as well as continued intrathecal therapy.
This backbone has been adopted by many groups, including the COG. Variation of this backbone includes the following: - Intensification for higher-risk patients by including additional doses of vincristine and pegaspargase, as well as repeated interim maintenance and delayed intensification phases.[2,3]
- Elimination or truncation of some of the phases for lower-risk patients to minimize acute and long-term toxicity.
Other clinical trial groups utilize a different therapeutic backbone during postinduction treatment phases: - DFCI: The DFCI ALL Consortium protocols include 20 to 30 weeks of pegaspargase therapy beginning at week 7 of therapy, given in conjunction with maintenance regimen (vincristine/dexamethasone pulses, low-dose methotrexate, nightly mercaptopurine).[4] These protocols also do not include a delayed intensification phase, but high-risk patients receive additional doses of doxorubicin (instead of methotrexate) during intensification.
- SJCRH: SJCRH follows a BFM backbone but augments the reinduction and maintenance phases for some patients by including intensified dosing of pegaspargase, frequent vincristine/corticosteroid pulses, and rotating drug pairs during maintenance.[5]
Standard-risk ALL In children with standard-risk B-cell ALL, there has been an attempt to limit exposure to drugs such as anthracyclines and alkylating agents that may be associated with an increased risk of late toxic effects.[6,7,8] For regimens utilizing a BFM backbone (such as COG), a single reinduction/delayed intensification phase, given with interim maintenance phases consisting of escalating doses of methotrexate (without leucovorin rescue) and vincristine, have been associated with favorable outcomes.[9] Favorable outcomes for standard-risk patients have also been reported by the Pediatric Oncology Group (POG), utilizing a limited number of courses of intermediate-dose or high-dose methotrexate as consolidation followed by maintenance therapy (without a reinduction phase),[7,10,11] and by the DFCI ALL Consortium utilizing multiple doses of pegaspargase (20-30 weeks) as consolidation, without postinduction exposure to alkylating agents or anthracyclines.[12,13] However, the prognostic impact of end-induction and/or consolidation minimal residual disease (MRD) has influenced the treatment of patients originally diagnosed as National Cancer Institute (NCI) standard risk. Multiple studies have demonstrated that higher levels of end-induction MRD are associated with poorer prognosis.[14,15,16,17,18] Augmenting therapy has been shown to improve the outcome in standard-risk patients with elevated MRD levels at the end of induction.[19] Therefore, standard-risk patients with higher levels of end-induction MRD are not treated with the approaches described for standard-risk patients who have low end-induction MRD, but are usually treated with high-risk regimens. Evidence (intensification for standard-risk ALL): - Clinical trials conducted in the 1980s and early 1990s demonstrated that the use of a delayed intensification phase improved outcome for children with standard-risk ALL treated with regimens using a BFM backbone.[20,21,22] The delayed intensification phase on such regimens, including those of the COG, consists of an 8-week phase of reinduction (including anthracycline) and reconsolidation containing cyclophosphamide, cytarabine, and 6-thioguanine given approximately 4 to 6 months after remission is achieved.[20,23,24]
- A Children's Cancer Group study (CCG-1991/COG-1991) for standard-risk ALL utilized dexamethasone in a three-drug induction phase and tested the utility of a second delayed intensification phase. This study also compared escalating intravenous (IV) methotrexate (without leucovorin rescue) in conjunction with vincristine versus a standard maintenance combination with oral methotrexate given during two interim maintenance phases.[9][Level of evidence: 1iiDi]
- A second delayed intensification phase provided no benefit in patients who were rapid early responders (M1 or M2 marrow by day 14 of induction).
- Escalating IV methotrexate during the interim maintenance phases, compared with oral methotrexate during these phases, produced a significant improvement in event-free survival (EFS), which was because of a decreased incidence of isolated extramedullary relapses, particularly those involving the CNS.
- In a randomized study conducted in the United Kingdom, children and young adults with ALL who lacked high-risk features (including adverse cytogenetics, and/or M3 marrow morphology at day 8 or day 15 of induction) were risk-stratified based on MRD level at the end of induction (week 4) and at week 11 of therapy. Patients with undetectable MRD at week 4 (or with low MRD at week 4 and undetectable by week 11) were considered low-risk, and were eligible to be randomly assigned to therapy with either one or two delayed intensification phases.[25][Level of evidence: 1iiDi]
- There was no significant difference in EFS between patients who received one and those who received two delayed intensification phases.
- There was no significant difference in treatment-related deaths between the two arms; however, the second delayed intensification phase was associated with grade 3 or 4 toxic events in 17% of the 261 patients randomly assigned to that arm, and one patient experienced a treatment-related death during that phase.
- Patients who are standard or intermediate risk at diagnosis, but have high levels of end-induction MRD, have been shown to have a poorer prognosis and should be treated as high-risk patients. The UKALL2003 (NCT00222612) trial used augmented postinduction therapy (extra doses of pegaspargase and vincristine and an escalated-dose of IV methotrexate without leucovorin rescue) to treat standard- or intermediate-risk patients with high levels of end-induction MRD.[19][Level of evidence: 1iiDi]
- Augmented postinduction therapy increased EFS to be comparable to that of patients with low levels of end-induction MRD.
High-risk ALL In high-risk patients, a number of different approaches have been used with comparable efficacy.[12,26]; [24][Level of evidence: 2Di] Treatment for high-risk patients generally is more intensive than that for standard-risk patients and typically includes higher cumulative doses of multiple agents, including anthracyclines and/or alkylating agents. Higher doses of these agents increase the risk of both short-term and long-term toxicities, and many clinical trials have focused on reducing the side effects of these intensified regimens. Evidence (intensification for high-risk ALL): - The former CCG developed an augmented BFM treatment regimen that included a second interim maintenance and delayed intensification phase. This regimen featured repeated courses of escalating-dose IV methotrexate (without leucovorin rescue) given with vincristine and pegaspargase during interim maintenance and additional vincristine and pegaspargase pulses during initial consolidation and delayed intensification. In the CCG-1882 trial, NCI high-risk patients with slow early response (M3 marrow on day 7 of induction) were randomly assigned to receive either standard- or augmented-BFM therapy.[2]
- The augmented therapy regimen in the CCG-1882 trial produced a significantly better EFS than did standard CCG modified BFM therapy.
- There was a significantly higher incidence of osteonecrosis in patients older than 10 years who received the augmented therapy (which included two 21-day postinduction dexamethasone courses), compared with those who were treated on the standard arm (one 21-day postinduction dexamethasone course).[27]
- In an Italian study, investigators showed that two applications of delayed intensification therapy (protocol II) significantly improved outcome for patients with a poor response to a prednisone prophase.[28]
- The CCG-1961 study used a 2 × 2 factorial design to compare both standard- versus augmented-intensity therapies and therapies of standard duration (one interim maintenance and delayed intensification phase) versus increased duration (two interim maintenance and delayed intensification phases) among NCI high-risk patients with a rapid early response. This trial also tested whether continuous versus alternate-week dexamethasone during delayed intensification phases affected rates of osteonecrosis.
- Augmented therapy was associated with an improvement in EFS; there was no EFS benefit associated with the administration of the second interim maintenance and delayed intensification phases.[3][Level of evidence: 1iiA]
- The cumulative incidence of osteonecrosis at 5 years was 9.9% for patients aged 10 to 15 years and 20.0% for patients aged 16 to 21 years, compared with 1.0% for patients aged 1 to 9 years (P = .0001). For patients aged 10 to 21 years, alternate-week dosing of dexamethasone during delayed intensification phases was associated with a significantly lower cumulative incidence of osteonecrosis, compared with continuous dosing (8.7% vs. 17.0%, P = .0005).[29][Level of evidence: 1iiC]
- In the COG AALL0232 (NCT00075725) study (2004-2011), patients with high-risk B-cell ALL received an augmented BFM backbone with one interim maintenance and delayed intensification phase; only patients with end-induction MRD greater than 0.1% or M2/M3 marrow at day 15 received two interim maintenance/delayed intensification phases. Patients were randomly assigned to receive either high-dose methotrexate or escalating dose IV methotrexate (Capizzi methotrexate) during the interim maintenance phase (the first phase only for those receiving two of these phases).[30,31]
- The methotrexate randomization was terminated early when planned interim monitoring indicated that high-dose methotrexate was associated with superior outcome. The 5-year EFS of patients randomly assigned to high-dose methotrexate was 79.6%, compared with 75% for those randomly assigned to the Capizzi methotrexate arm. High-dose methotrexate was also associated with a superior 5-year overall survival (OS) (P = .025).[31]
- Patients with MRD less than 0.01% at end of induction had a 5-year EFS of 87%, compared with 74% for those with MRD 0.01% to 0.1%. Those with MRD levels greater than 0.1% fared worse.[30]
- High-dose methotrexate was associated with a superior EFS in patients with end-induction MRD greater than 0.01% (high-dose methotrexate, 68%; Capizzi methotrexate, 58%; P = .008).[30]
Because treatment for high-risk ALL involves more intensive therapy, leading to a higher risk of acute and long-term toxicities, a number of clinical trials have tested interventions to prevent side effects without adversely impacting EFS. Interventions that have been investigated include the use of the cardioprotectant dexrazoxane (to prevent anthracycline-related cardiac toxic effects) and alternative scheduling of corticosteroids (to reduce the risk of osteonecrosis). Evidence (cardioprotective effect of dexrazoxane): - In a DFCI ALL Consortium trial, children with high-risk ALL were randomly assigned to receive doxorubicin alone (30 mg/m2 /dose to a cumulative dose of 300 mg/m2) or the same dose of doxorubicin with dexrazoxane during the induction and intensification phases of multiagent chemotherapy.[32,33]
- The use of the cardioprotectant dexrazoxane before doxorubicin resulted in better left ventricular fractional shortening and improved end-systolic dimension Z-scores without any adverse effect on EFS or increase in second malignancy risk, compared with the use of doxorubicin alone 5 years posttreatment.
- A greater long-term protective effect was noted in girls than in boys.
- On the POG-9404 trial, patients with T-cell ALL were randomly assigned to receive dexrazoxane or not before each dose of doxorubicin (cumulative dose 360 mg/m2).[34]
- There was no difference in EFS between patients with T-cell ALL who were treated with dexrazoxane and patients who were not treated with dexrazoxane (cumulative doxorubicin dose, 360 mg/m2).
- The frequency of grade 3 and 4 toxicities that occurred during therapy was similar between the randomized groups, and there was no difference in cumulative incidence of second malignant neoplasms. Three years after initial diagnosis, left ventricular shortening fraction and left ventricular wall thickness were both significantly worse in patients who received doxorubicin alone than in patients who received dexrazoxane, indicating that dexrazoxane was cardioprotective.
Evidence (reducing risk of osteonecrosis): - In the CCG-1961 study, alternate-week dosing of dexamethasone during delayed intensification was studied with the goal of reducing the frequency of osteonecrosis.[29] On that protocol, patients with high-risk B-cell ALL and a rapid early morphologic response to induction therapy were randomly assigned to receive either one or two delayed intensification phases. Those randomly assigned to one delayed intensification phase received daily dosing of dexamethasone (21 consecutive days), while those randomly assigned to two delayed intensification phases received alternate-week dosing of dexamethasone (days 0-6 and 14-21) during each delayed intensification phase.
- For patients aged 10 years or older at diagnosis, those who received two delayed intensification phases (alternate-week dosing of dexamethasone) had a significantly lower risk of symptomatic osteonecrosis (5-year cumulative incidence of 8.7%, compared with 17% for patients receiving one delayed intensification phase with continuous dexamethasone dosing; P = .001).
- The greatest impact was seen in females aged 16 to 21 years, who showed the highest incidence of osteonecrosis with standard therapy containing continuous dexamethasone; the incidence of osteonecrosis with alternative-week dexamethasone was 5.6%, compared with 57.6% for those receiving continuous dosing.
(Refer to the Osteonecrosis section of this summary for more information.) Very high-risk ALL Approximately 10% to 20% of patients with ALL are classified as very high risk, including the following:[24,35] - Infants younger than 1 year, especially if there is an MLL (KMT2A) gene rearrangement present. (Refer to the Infants With ALL subsection in the Postinduction Treatment for Specific ALL Subgroups section of this summary for more information about infants with ALL.)
- Patients with adverse cytogenetic abnormalities, including t(9;22)(q34;q11.2), t(17;19), MLL gene rearrangements, and low hypodiploidy (<44 chromosomes).
- Patients who achieve CR but have a slow early response to initial therapy, including those with a high absolute blast count after a 7-day steroid prophase, and patients with high MRD levels at the end of induction (week 4) or later time points (e.g., week 12).
- Patients who have morphologically persistent disease after the first 4 weeks of therapy (induction failure), even if they later achieve CR.
COG also considers patients who are aged 13 years or older to be very high risk, although this age criterion is not utilized by other groups. Patients with very high-risk features have been treated with multiple cycles of intensive chemotherapy during the consolidation phase (usually in addition to the typical BFM backbone intensification phases). These additional cycles often include agents not typically used in frontline ALL regimens for standard-risk and high-risk patients, such as high-dose cytarabine, ifosfamide, and etoposide.[24] However, even with this intensified approach, reported long-term EFS rates range from 30% to 50% for this patient subset.[24,36] On some clinical trials, very high-risk patients have also been considered candidates for allogeneic hematopoietic stem cell transplantation (HSCT) in first CR.[36,37,38,39] However, there are limited data regarding the outcome of very high-risk patients treated with allogeneic HSCT in first CR. Controversy exists regarding which subpopulations could potentially benefit from HSCT. Evidence (allogeneic HSCT in first remission for very high-risk patients): - In a European cooperative group study conducted between 1995 and 2000, very high-risk patients (defined as one of the following: morphologically persistent disease after a four-drug induction, t(9;22)(q34;q11.2) or t(4;11)(q21;q23), or poor response to prednisone prophase in patients with either T-cell phenotype or presenting white blood cells [WBC] >100,000/μL) were assigned to receive either an allogeneic HSCT in first CR (based on the availability of a human lymphocyte antigen-matched related donor) or intensive chemotherapy.[36]
- Using an intent-to-treat analysis, patients assigned to allogeneic HSCT (on the basis of donor availability) had a superior 5-year disease-free survival (DFS) compared with patients assigned to intensive chemotherapy (57% ± 7% for transplant vs. 41% ± 3% for chemotherapy, P = .02)
- There was no significant difference in OS (56% ± 6% for transplant vs. 50% ± 3% for chemotherapy, P = .12).
- For patients with T-cell ALL and a poor response to prednisone prophase, both DFS and OS rates were significantly better with allogeneic HSCT.[37]
- In a large retrospective series of patients with initial induction failure, the 10-year OS for patients with persistent leukemia was 32%.[40]
- A trend for superior outcome with allogeneic HSCT, compared with chemotherapy alone, was observed in patients with T-cell phenotype (any age) and with precursor B-cell ALL who were older than 6 years.
- Patients with precursor B-cell ALL who were aged 1 to 5 years at diagnosis and did not have any adverse cytogenetic abnormalities (MLL [KMT2A] rearrangement, BCR-ABL1) had a relatively favorable prognosis, without any advantage in outcome with the utilization of HSCT compared with chemotherapy alone.
- The Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) ALL-BFM-2000 (NCT00430118) study (2000-2006) classified patients as high risk if they met any of the following criteria: poor response to prednisone prophase, failure to achieve CR at the end of the first month of treatment, high MRD levels after induction IB (day 78 of therapy), and t(4;11)(q21;q23). These patients were allocated to allogeneic HSCT in first CR per protocol on the basis of donor availability and investigator preference.[41][Level of evidence: 2Dii]
- The overall 5-year EFS of patients meeting high-risk criteria was 58.9%.
- The 5-year EFS was 74% for patients whose only high-risk feature was prednisone-poor response; there was no significant difference in DFS (P = .31) or OS (P = .91) when comparing HSCT and chemotherapy for patients with poor prednisone response in whom HSCT was allowed per protocol (those with T-cell ALL and/or WBC ≥100,000/mm3).
- All other high-risk patients (i.e., those with initial induction failure, high day 78 MRD and/or t(4;11)(q21;q23)) had EFS rates less than 50%. For these patients, there was no statistically significant difference in DFS between those who received HSCT (n = 66) and those who received chemotherapy only (n = 88), after adjusting for waiting time to HSCT (5.7 months).
Maintenance therapy Backbone of maintenance therapy The backbone of maintenance therapy in most protocols includes daily oral mercaptopurine and weekly oral or parenteral methotrexate. Clinical practice generally calls for the administration of oral mercaptopurine in the evening, based on evidence from older studies that this practice may improve EFS.[42] On many protocols, intrathecal chemotherapy for CNS sanctuary therapy is continued during maintenance therapy. It is imperative to carefully monitor children on maintenance therapy for both drug-related toxicity and for compliance with the oral chemotherapy agents used during maintenance therapy.[43] Studies conducted by the COG have demonstrated significant differences in compliance with mercaptopurine (6-MP) amongst various racial and socioeconomic groups. Importantly, nonadherence to treatment with mercaptopurine in the maintenance phase was associated with a significant increase in the risk of relapse.[43,44] Some patients may develop severe hematologic toxicity when receiving conventional dosages of mercaptopurine because of an inherited deficiency (homozygous mutant) of thiopurine S-methyltransferase, an enzyme that inactivates mercaptopurine.[45,46] These patients are able to tolerate mercaptopurine only if dosages much lower than those conventionally used are administered.[45,46] Patients who are heterozygous for this mutant enzyme gene generally tolerate mercaptopurine without serious toxicity, but they do require more frequent dose reductions for hematologic toxicity than do patients who are homozygous for the normal allele.[45] Polymorphisms of the NUDT15 gene, observed most frequently in East Asian and Hispanic patients, have also been linked to extreme sensitivity to the myelosuppressive effects of mercaptopurine.[47,48] Evidence (maintenance therapy): - In a meta-analysis of randomized trials that compared thiopurines, thioguanine did not improve the overall EFS, although particular subgroups may benefit from its use.[49] The use of continuous thioguanine instead of mercaptopurine during the maintenance phase is associated with an increased risk of hepatic complications, including veno-occlusive disease and portal hypertension.[50,51,52,53,54] Because of the increased toxicity of thioguanine, mercaptopurine remains the standard drug of choice.
- An intensified maintenance regimen, consisting of rotating pairs of agents, including cyclophosphamide and epipodophyllotoxins along with more standard maintenance agents, has been evaluated in several clinical trials conducted by SJCRH and other groups.[55]
- The intensified maintenance with rotating pairs of agents has been associated with more episodes of febrile neutropenia [56] and a higher risk of secondary acute myelogenous leukemia,[57,58] especially when epipodophyllotoxins are included.[56]
Based on these findings, SJCRH has modified the agents used in the rotating pair schedule during the maintenance phase. On the Total XV study, standard-risk and high-risk patients received three rotating pairs (mercaptopurine plus methotrexate, cyclophosphamide plus cytarabine, and dexamethasone plus vincristine) throughout this treatment phase; low-risk patients received more standard maintenance (without cyclophosphamide and cytarabine).[5] - A randomized study from Argentina demonstrated no benefit from this intensified approach compared with a more standard maintenance regimen for patients who receive induction and consolidation phases based on a BFM backbone.[56]
Vincristine/corticosteroid pulses Pulses of vincristine and corticosteroid are often added to the standard maintenance backbone, although the benefit of these pulses within the context of contemporary multiagent chemotherapy regimens remains controversial. Evidence (vincristine/corticosteroid pulses): - A CCG randomized trial conducted in the 1980s demonstrated improved outcome in patients receiving monthly vincristine/prednisone pulses.[59]
- A meta-analysis combining data from six clinical trials from the same treatment era showed an EFS advantage for vincristine/prednisone pulses.[60,61]
- A systematic review of the impact of vincristine plus steroid pulses from more recent clinical trials raised the question of whether such pulses are of value in current ALL treatment, which includes more intensive early therapy.[61]
- In a multicenter randomized trial in children with intermediate-risk ALL being treated on a BFM regimen, there was no benefit associated with the addition of six pulses of vincristine/dexamethasone during the continuation phase, although the pulses were administered less frequently than in other trials in which a benefit had been demonstrated.[62]
- A small multicenter trial of average-risk patients demonstrated superior EFS in patients receiving vincristine plus corticosteroid pulses. In this study, there was no difference in outcome based on type of steroid (prednisone vs. dexamethasone).[63][Level of evidence: 1iiA]
For regimens that include vincristine/steroid pulses, a number of studies have addressed which steroid (dexamethasone or prednisone) should be used. From these studies, it appears that dexamethasone is associated with superior EFS, but also may lead to a greater frequency of steroid-associated complications, including bone toxicity and infections, especially in older children and adolescents. Dexamethasone has not been associated with an increased frequency of these complications in younger patients.[20,64,65,66,67] Evidence (dexamethasone vs. prednisone): - In a CCG study, dexamethasone was compared with prednisone for children aged 1 to younger than 10 years with lower-risk ALL.[20,64]
- Patients randomly assigned to receive dexamethasone had significantly fewer CNS relapses and a significantly better EFS rate.
- In an MRC UK-ALL trial, dexamethasone was compared with prednisolone during induction and maintenance therapies in both standard-risk and high-risk patients.[65]
- The EFS and incidence of both CNS and non-CNS relapses improved with the use of dexamethasone.
- Dexamethasone was associated with an increased risk of steroid-associated toxicities, including behavioral problems, myopathy, and osteopenia.
- In a DFCI ALL Consortium trial, patients were randomly assigned to receive either dexamethasone or prednisone during all postinduction treatment phases.[67]
- Dexamethasone was associated with a superior EFS, but also with a higher frequency of infections (primarily episodes of bacteremia) and, in patients aged 10 years or older, an increased incidence of osteonecrosis and fracture.
The benefit of using dexamethasone in children aged 10 to 18 years requires further investigation because of the increased risk of steroid-induced osteonecrosis in this age group.[27,66] Duration of maintenance therapy Maintenance chemotherapy generally continues until 2 to 3 years of continuous CR. On some studies, boys are treated longer than girls;[20] on others, there is no difference in the duration of treatment based on gender.[12,24] It is not clear whether longer duration of maintenance therapy reduces relapse in boys, especially in the context of current therapies.[24][Level of evidence: 2Di] Extending the duration of maintenance therapy beyond 3 years does not improve outcome.[60] Adherence to oral medications during maintenance therapy Nonadherence to treatment with mercaptopurine during maintenance therapy is associated with a significant risk of relapse.[43] Evidence (adherence to treatment): - The COG studied the impact of nonadherence to mercaptopurine during maintenance therapy in 327 children and adolescents (169 Hispanics and 158 non-Hispanic whites).[43]
- A progressive increase in relapse was observed with decreasing adherence to mercaptopurine, with hazard ratios (HRs) ranging between 4.0% to 5.7% for adherence rates ranging from 94.9% to 90%, 89.9% to 85%, and less than 85%. After adjusting for other prognostic factors (including NCI risk group and chromosomal abnormalities), a progressive increase in relapse was observed with decreasing adherence to mercaptopurine.
- Adherence was significantly lower among Hispanics, patients older than 12 years, and patients from single-mother households.
- A second study of adherence was conducted in 298 children with ALL (71 Asian Americans, 68 African Americans, and 159 non-Hispanic whites).[44]
- An adherence rate of less than 90% was associated with increased relapse risk (HR, 3.9).
- Using an adherence rate of less than 90% to define nonadherence, 20.5% of the participants were nonadherers.
- Adherence rates were significantly lower in Asian Americans and African Americans than in non-Hispanic whites.
- In a third study of 742 children, the following key observations were made:[68]
- Patients with mercaptopurine nonadherence (defined as mean adherence rate < 95%) were at 2.7-fold increased risk of relapse compared to adherers.
- Amongst adherers, high intra-individual variability in thioguanine levels (due to varying dose-intensity and drug treatment interruptions) was associated with increased risk of relapse.
- The authors of the above studies also found that self-reporting was not a reliable measure of adherence, with 84% of patients over reporting compliance with taking mercaptopurine at least some of the time.[69] The data suggest that additional measures of adherence besides self-reporting are needed.
Treatment options under clinical evaluation Risk-based treatment assignment is a key therapeutic strategy utilized for children with ALL, and protocols are designed for specific patient populations that have varying degrees of risk of treatment failure. The Risk-Based Treatment Assignment section of this summary describes the clinical and laboratory features used for the initial stratification of children with ALL into risk-based treatment groups. Ongoing clinical trials include the following: COG studies for precursor B-cell ALL Standard-risk ALL - COG-AALL0932 (NCT01190930) (Risk-Adapted Chemotherapy in Younger Patients With Newly Diagnosed Standard-Risk ALL):
All patients will receive a three-drug induction (dexamethasone, vincristine, and IV pegaspargase) with intrathecal chemotherapy. Postinduction therapeutic questions have been answered for low-risk and average-risk patients, and trials have met accrual goals and are closed; protocol therapy ends after the first month of therapy for all patients, except for those with Down syndrome who have low bone marrow MRD on day 29. The study objective is to describe the outcome of standard-risk Down syndrome patients treated with a standardized treatment and enhanced supportive care. Non-Down syndrome patients on this study who are found to have high-risk features are eligible to enroll on COG-AALL1131 after induction.
High-risk and very high-risk ALL - COG-AALL1131 (NCT01406756) (Combination Chemotherapy in Treating Young Patients With Newly Diagnosed High-Risk ALL):
This protocol is open to B-cell ALL patients aged 30 years or younger. Patients on this trial are classified as high risk if they are: - NCI high risk (by age or WBC) but lack very high-risk features (see below); or
- NCI standard risk who lack very high-risk features and meet either of the following criteria:
- Absence of favorable genetics (no ETV6-RUNX1 or double trisomies 4 and 10) and day 8 peripheral blood MRD greater than 1%.
- Presence of favorable cytogenetics and high marrow MRD at day 29.
Patients are classified as very high risk if they meet any of the following criteria: - Age 13 years and older.
- CNS3 at diagnosis.
- M3 marrow at day 29.
- Unfavorable genetics (e.g., iAMP21, low hypodiploidy, MLL [KMT2A] gene rearrangement).
- High marrow MRD (> 0.01% by flow cytometry) at day 29 (with the exception of NCI standard-risk patients with favorable genetics).
Patients on this trial receive a four-drug induction (vincristine, corticosteroid, daunorubicin, and IV pegaspargase) with intrathecal chemotherapy. Patients younger than 10 years receive 2 weeks of dexamethasone during induction, and those aged 10 years and older receive 4 weeks of prednisone. For high-risk patients, postinduction therapy consists of a modified augmented BFM backbone, including an interim maintenance phase with high-dose methotrexate and one delayed intensification phase. The study compares triple intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) with intrathecal methotrexate in a randomized fashion to determine whether triple intrathecal chemotherapy reduces CNS relapse rates and improves EFS. Patients identified as having BCR-ABL1-like ALL and who have a gene fusion involving a kinase that is sensitive to dasatinib (e.g., ABL1, ABL2, CSF1F, and PDGFRB) are assigned to treatment with dasatinib added to standard chemotherapy. Dasatinib treatment is initiated after induction therapy is complete, and it continues through maintenance therapy. For very high-risk patients, the study had evaluated whether intensification of the consolidation phase and second-half of delayed intensification phases improved DFS. However, that portion of the trial was closed when a futility boundary was crossed, indicating that the study would not be able to demonstrate the superiority of the experimental arm. Therefore, very high-risk patients are removed from protocol therapy at the end of induction. - COG-AALL1521 (NCT02723994) (A Phase 2 Study of Ruxolitinib With Chemotherapy in Children With ALL): This nonrandomized study is testing the addition of ruxolitinib (JAK-inhibitor) in combination with the modified augmented BFM regimen (similar to AALL1131) for the treatment of NCI high-risk B-cell ALL (ages 1-21 years) with any of the following genetic abnormalities: 1) rearranged CRLF2 (CRLF-R); 2) mutations in JAK1 or JAK2; or 3) other alterations involving the JAK pathway (e.g., JAK2 fusions, EPO-R fusions, SH2B3 deletions, IL7RA mutations). Patients enter the study after completing the induction phase. Ruxolitinib will be administered in conjunction with all postinduction treatment phases. The primary objective is to evaluate the safety, tolerability, and efficacy of the combination.
Other studies - Total XVI study (TOTXVI) (Total Therapy Study XVI for Newly Diagnosed Patients With ALL): A study at SJCRH is randomly assigning patients to receive either standard-dose (2,500 u/m2) or high-dose (3,500 u/m2) pegaspargase during postremission therapy.
- DFCI ALL 16-001 (NCT03020030) (Risk Classification Schemes in Identifying Better Treatment Options for Children and Adolescents with ALL): This trial has two main objectives: 1) To test a novel risk classification scheme for children and adolescents with ALL, and 2) to test the feasibility of administering pegaspargase at a reduced dose during postinduction treatment phases (adjusting doses based on serum asparaginase activity levels), with the goal of maintaining therapeutic serum asparaginase activity levels while potentially reducing nonallergic asparaginase-related toxicities.
Patients are assigned an initial risk group by day 10 of therapy. Patients are considered initial very high risk if any of the following are present: IKZF1 deletion, MLL gene rearrangement, or low hypodiploidy (<40 chromosomes). Patients are considered initial low risk if they meet all of the following criteria: B-cell ALL, aged 1 year to younger than 15 years, WBC count less than 50 × 109, CNS1 or CNS2, absence of iAMP21, and absence of very high-risk features. Initial high-risk patients include all other patients lacking very high-risk features, including all patients with T-cell ALL. Intensity of induction depends on initial risk group. Initial low-risk patients receive a three-drug induction (no anthracycline). All other patients receive a four-drug induction (with anthracycline). Final risk group, which determines the intensity of postinduction therapy, is assigned based on MRD (assessed by next-generation sequencing) at the end of induction (day 32; first time point) and week 10 (second time point). - Initial low-risk patients with low MRD (<104) at the first time point are considered final low risk. They continue treatment per DFCI standard-risk backbone, including 30 weeks of pegaspargase, but without any anthracycline.
- Initial low-risk patients with high MRD (>104) at the first time point but low MRD (<103) at the second time point and all initial high-risk patients with low MRD (<103) at the second time point continue treatment per DFCI high-risk backbone, including doxorubicin, but with a reduced dose of dexamethasone compared with previous trials.
- All initial very high-risk patients and any initial low-risk/high-risk patient with high MRD (>103) at the second time point are considered very high risk and receive an intensified consolidation phase followed by the DFCI high-risk backbone. Any very high-risk patients identified as having BCR-ABL1-like ALL with a gene fusion involving a kinase that is sensitive to dasatinib (e.g., ABL1, ABL2, CSF1F, and PDGFRB) will receive dasatinib during all postinduction treatment phases.
Treatment for all risk groups includes 30 weeks of pegaspargase (15 doses given every 2 weeks) during postinduction therapy. All final low-risk/high-risk patients are eligible to participate in a randomized comparison of postinduction pegaspargase dosing: standard dose (2,500 IU/m2 /dose) or pharmacokinetic-adjusted reduced dose (starting dose: 2,000 IU/m2). In all patients, nadir serum asparaginase activity (NSAA) is checked before each pegaspargase dose; any patient found to have a nondetectable NSAA is switched to Erwinia asparaginase. On the pharmacokinetic-adjusted reduced-dose arm, the dose may be decreased further to 1,750 IU/m2 if NSAA is found to be extremely high (>1.0 IU/mL) after the fourth pegaspargase dose; the dose will be increased up to standard dose (2,500 IU/m2) if NSAA is low but detectable (<0.4 IU/mL) at any time point. The trial is also piloting a strategy to rechallenge patients with grade 2 hypersensitivity reactions to pegaspargase with pharmacokinetic-monitoring to determine whether such patients will switch to Erwinia or may continue to receive pegaspargase with premedication.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with childhood acute lymphoblastic leukemia in remission. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
-
Nachman JB, Sather HN, Sensel MG, et al.: Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 338 (23): 1663-71, 1998.
-
Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.
-
Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
-
Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
-
Veerman AJ, Hählen K, Kamps WA, et al.: High cure rate with a moderately intensive treatment regimen in non-high-risk childhood acute lymphoblastic leukemia. Results of protocol ALL VI from the Dutch Childhood Leukemia Study Group. J Clin Oncol 14 (3): 911-8, 1996.
-
Chauvenet AR, Martin PL, Devidas M, et al.: Antimetabolite therapy for lesser-risk B-lineage acute lymphoblastic leukemia of childhood: a report from Children's Oncology Group Study P9201. Blood 110 (4): 1105-11, 2007.
-
Gustafsson G, Kreuger A, Clausen N, et al.: Intensified treatment of acute childhood lymphoblastic leukaemia has improved prognosis, especially in non-high-risk patients: the Nordic experience of 2648 patients diagnosed between 1981 and 1996. Nordic Society of Paediatric Haematology and Oncology (NOPHO) Acta Paediatr 87 (11): 1151-61, 1998.
-
Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
-
Mahoney DH Jr, Shuster JJ, Nitschke R, et al.: Intensification with intermediate-dose intravenous methotrexate is effective therapy for children with lower-risk B-precursor acute lymphoblastic leukemia: A Pediatric Oncology Group study. J Clin Oncol 18 (6): 1285-94, 2000.
-
Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
-
Silverman LB, Gelber RD, Dalton VK, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97 (5): 1211-8, 2001.
-
Pession A, Valsecchi MG, Masera G, et al.: Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J Clin Oncol 23 (28): 7161-7, 2005.
-
Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
-
van Dongen JJ, Seriu T, Panzer-Grümayer ER, et al.: Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 352 (9142): 1731-8, 1998.
-
Zhou J, Goldwasser MA, Li A, et al.: Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood 110 (5): 1607-11, 2007.
-
Coustan-Smith E, Sancho J, Hancock ML, et al.: Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood 100 (7): 2399-402, 2002.
-
Stow P, Key L, Chen X, et al.: Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood 115 (23): 4657-63, 2010.
-
Vora A, Goulden N, Mitchell C, et al.: Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol 15 (8): 809-18, 2014.
-
Gaynon PS, Angiolillo AL, Carroll WL, et al.: Long-term results of the children's cancer group studies for childhood acute lymphoblastic leukemia 1983-2002: a Children's Oncology Group Report. Leukemia 24 (2): 285-97, 2010.
-
Riehm H, Gadner H, Henze G, et al.: Results and significance of six randomized trials in four consecutive ALL-BFM studies. Hamatol Bluttransfus 33: 439-50, 1990.
-
Hutchinson RJ, Gaynon PS, Sather H, et al.: Intensification of therapy for children with lower-risk acute lymphoblastic leukemia: long-term follow-up of patients treated on Children's Cancer Group Trial 1881. J Clin Oncol 21 (9): 1790-7, 2003.
-
Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
-
Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
-
Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
-
Pui CH, Mahmoud HH, Rivera GK, et al.: Early intensification of intrathecal chemotherapy virtually eliminates central nervous system relapse in children with acute lymphoblastic leukemia. Blood 92 (2): 411-5, 1998.
-
Mattano LA Jr, Sather HN, Trigg ME, et al.: Osteonecrosis as a complication of treating acute lymphoblastic leukemia in children: a report from the Children's Cancer Group. J Clin Oncol 18 (18): 3262-72, 2000.
-
Aricò M, Valsecchi MG, Conter V, et al.: Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisone-poor response treated with double Berlin-Frankfurt-Muenster protocol II. Blood 100 (2): 420-6, 2002.
-
Mattano LA Jr, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.
-
Borowitz MJ, Wood BL, Devidas M, et al.: Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood 126 (8): 964-71, 2015.
-
Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.
-
Lipshultz SE, Scully RE, Lipsitz SR, et al.: Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol 11 (10): 950-61, 2010.
-
Barry EV, Vrooman LM, Dahlberg SE, et al.: Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J Clin Oncol 26 (7): 1106-11, 2008.
-
Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.
-
Schultz KR, Pullen DJ, Sather HN, et al.: Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood 109 (3): 926-35, 2007.
-
Balduzzi A, Valsecchi MG, Uderzo C, et al.: Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: comparison by genetic randomisation in an international prospective study. Lancet 366 (9486): 635-42, 2005 Aug 20-26.
-
Schrauder A, Reiter A, Gadner H, et al.: Superiority of allogeneic hematopoietic stem-cell transplantation compared with chemotherapy alone in high-risk childhood T-cell acute lymphoblastic leukemia: results from ALL-BFM 90 and 95. J Clin Oncol 24 (36): 5742-9, 2006.
-
Ribera JM, Ortega JJ, Oriol A, et al.: Comparison of intensive chemotherapy, allogeneic, or autologous stem-cell transplantation as postremission treatment for children with very high risk acute lymphoblastic leukemia: PETHEMA ALL-93 Trial. J Clin Oncol 25 (1): 16-24, 2007.
-
Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
-
Schrappe M, Hunger SP, Pui CH, et al.: Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 366 (15): 1371-81, 2012.
-
Conter V, Valsecchi MG, Parasole R, et al.: Childhood high-risk acute lymphoblastic leukemia in first remission: results after chemotherapy or transplant from the AIEOP ALL 2000 study. Blood 123 (10): 1470-8, 2014.
-
Schmiegelow K, Glomstein A, Kristinsson J, et al.: Impact of morning versus evening schedule for oral methotrexate and 6-mercaptopurine on relapse risk for children with acute lymphoblastic leukemia. Nordic Society for Pediatric Hematology and Oncology (NOPHO). J Pediatr Hematol Oncol 19 (2): 102-9, 1997 Mar-Apr.
-
Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
-
Bhatia S, Landier W, Hageman L, et al.: 6MP adherence in a multiracial cohort of children with acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 124 (15): 2345-53, 2014.
-
Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
-
Andersen JB, Szumlanski C, Weinshilboum RM, et al.: Pharmacokinetics, dose adjustments, and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr 87 (1): 108-11, 1998.
-
Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
-
Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
-
Escherich G, Richards S, Stork LC, et al.: Meta-analysis of randomised trials comparing thiopurines in childhood acute lymphoblastic leukaemia. Leukemia 25 (6): 953-9, 2011.
-
Broxson EH, Dole M, Wong R, et al.: Portal hypertension develops in a subset of children with standard risk acute lymphoblastic leukemia treated with oral 6-thioguanine during maintenance therapy. Pediatr Blood Cancer 44 (3): 226-31, 2005.
-
De Bruyne R, Portmann B, Samyn M, et al.: Chronic liver disease related to 6-thioguanine in children with acute lymphoblastic leukaemia. J Hepatol 44 (2): 407-10, 2006.
-
Vora A, Mitchell CD, Lennard L, et al.: Toxicity and efficacy of 6-thioguanine versus 6-mercaptopurine in childhood lymphoblastic leukaemia: a randomised trial. Lancet 368 (9544): 1339-48, 2006.
-
Jacobs SS, Stork LC, Bostrom BC, et al.: Substitution of oral and intravenous thioguanine for mercaptopurine in a treatment regimen for children with standard risk acute lymphoblastic leukemia: a collaborative Children's Oncology Group/National Cancer Institute pilot trial (CCG-1942). Pediatr Blood Cancer 49 (3): 250-5, 2007.
-
Stork LC, Matloub Y, Broxson E, et al.: Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children's Oncology Group CCG-1952 clinical trial. Blood 115 (14): 2740-8, 2010.
-
Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
-
Felice MS, Rossi JG, Gallego MS, et al.: No advantage of a rotational continuation phase in acute lymphoblastic leukemia in childhood treated with a BFM back-bone therapy. Pediatr Blood Cancer 57 (1): 47-55, 2011.
-
Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.
-
Pui CH, Ribeiro RC, Hancock ML, et al.: Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med 325 (24): 1682-7, 1991.
-
Bleyer WA, Sather HN, Nickerson HJ, et al.: Monthly pulses of vincristine and prednisone prevent bone marrow and testicular relapse in low-risk childhood acute lymphoblastic leukemia: a report of the CCG-161 study by the Childrens Cancer Study Group. J Clin Oncol 9 (6): 1012-21, 1991.
-
Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomised children. Childhood ALL Collaborative Group. Lancet 347 (9018): 1783-8, 1996.
-
Eden TO, Pieters R, Richards S, et al.: Systematic review of the addition of vincristine plus steroid pulses in maintenance treatment for childhood acute lymphoblastic leukaemia - an individual patient data meta-analysis involving 5,659 children. Br J Haematol 149 (5): 722-33, 2010.
-
Conter V, Valsecchi MG, Silvestri D, et al.: Pulses of vincristine and dexamethasone in addition to intensive chemotherapy for children with intermediate-risk acute lymphoblastic leukaemia: a multicentre randomised trial. Lancet 369 (9556): 123-31, 2007.
-
De Moerloose B, Suciu S, Bertrand Y, et al.: Improved outcome with pulses of vincristine and corticosteroids in continuation therapy of children with average risk acute lymphoblastic leukemia (ALL) and lymphoblastic non-Hodgkin lymphoma (NHL): report of the EORTC randomized phase 3 trial 58951. Blood 116 (1): 36-44, 2010.
-
Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.
-
Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.
-
Strauss AJ, Su JT, Dalton VM, et al.: Bony morbidity in children treated for acute lymphoblastic leukemia. J Clin Oncol 19 (12): 3066-72, 2001.
-
Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
-
Bhatia S, Landier W, Hageman L, et al.: Systemic Exposure to Thiopurines and Risk of Relapse in Children With Acute Lymphoblastic Leukemia: A Children's Oncology Group Study. JAMA Oncol 1 (3): 287-95, 2015.
-
Landier W, Chen Y, Hageman L, et al.: Comparison of self-report and electronic monitoring of 6MP intake in childhood ALL: a Children's Oncology Group study. Blood 129 (14): 1919-1926, 2017.
CNS-Directed Therapy for Childhood ALLAt diagnosis, approximately 3% of patients have central nervous system 3 (CNS3) disease (defined as cerebrospinal fluid [CSF] specimen with ≥5 white blood cells [WBC]/μL with lymphoblasts and/or the presence of cranial nerve palsies). However, unless specific therapy is directed toward the CNS, most children will eventually develop overt CNS leukemia whether or not lymphoblasts were detected in the spinal fluid at initial diagnosis. Therefore, all children with acute lymphoblastic leukemia (ALL) should receive systemic combination chemotherapy together with some form of CNS prophylaxis. Because the CNS is a sanctuary site (i.e., an anatomic space that is poorly penetrated by many of the systemically administered chemotherapy agents typically used to treat ALL), specific CNS-directed therapies must be instituted early in treatment to eliminate clinically evident CNS disease at diagnosis and to prevent CNS relapse in all patients. Historically, survival rates for children with ALL improved dramatically after CNS-directed therapies were added to treatment regimens. Standard treatment options for CNS-directed therapy include the following: - Intrathecal chemotherapy.
- CNS-directed systemic chemotherapy.
- Cranial radiation.
All of these treatment modalities have a role in the treatment and prevention of CNS leukemia. The combination of intrathecal chemotherapy plus CNS-directed systemic chemotherapy is standard; cranial radiation is reserved for selective situations.[1] The type of CNS-therapy that is used is based on a patient's risk of CNS-relapse, with higher-risk patients receiving more intensive treatments. Data suggest that the following groups of patients are at increased risk of CNS relapse: - Patients with 5 or more WBC/µL and blasts in the CSF (CNS3), obtained at diagnosis.
- Patients with blasts in the CSF but fewer than 5 WBC/µL (CNS2) may be at increased risk of CNS relapse,[2] although this risk appears to be nearly fully abrogated if they receive more doses of intrathecal chemotherapy, especially during the induction phase.[3]
- Patients with T-cell ALL, especially those with high presenting peripheral blood leukocyte counts.
- Patients who have a traumatic lumbar puncture showing blasts at the time of diagnosis may have an increased risk of CNS relapse. These patients receive more intensive CNS-directed therapy on some treatment protocols.[3,4]
CNS-directed treatment regimens for newly diagnosed childhood ALL are presented in Table 2: Table 2. CNS-Directed Treatment Regimens for Newly Diagnosed Childhood ALL| Disease Status | Standard Treatment Options |
|---|
| ALL = acute lymphoblastic leukemia; CNS = central nervous system; CNS3 = cerebrospinal fluid with five or more white blood cells/µL, cytospin positive for blasts, or cranial nerve palsies. | | a The drug itself is not CNS-penetrant, but leads to cerebrospinal fluid asparagine depletion. | | Standard-risk ALL | Intrathecal chemotherapy | | | Methotrexate alone | | | Methotrexate with cytarabine and hydrocortisone | | CNS-directed systemic chemotherapy | | | Dexamethasone | | | L-asparaginasea | | | High-dose methotrexate with leucovorin rescue | | | Escalating-dose intravenous methotrexate (no leucovorin rescue) | | High-risk and very high-risk ALL | Intrathecal chemotherapy | | | Methotrexate alone | | | Methotrexate with cytarabine and hydrocortisone | | CNS-directed systemic chemotherapy | | | Dexamethasone | | | L-asparaginasea | | | High-dose methotrexate with leucovorin rescue | | Cranial radiation | A major goal of current ALL clinical trials is to provide effective CNS therapy while minimizing neurologic toxic effects and other late effects. Intrathecal Chemotherapy All therapeutic regimens for childhood ALL include intrathecal chemotherapy. Intrathecal chemotherapy is usually started at the beginning of induction, intensified during consolidation and, in many protocols, continued throughout the maintenance phase. Intrathecal chemotherapy typically consists of one of the following:[5] - Methotrexate alone.
- Methotrexate with cytarabine and hydrocortisone (triple intrathecal chemotherapy).
Unlike intrathecal cytarabine, intrathecal methotrexate has a significant systemic effect, which may contribute to prevention of marrow relapse.[6] CNS-Directed Systemic Chemotherapy In addition to therapy delivered directly to the brain and spinal fluid, systemically administered agents are also an important component of effective CNS prophylaxis. The following systemically administered drugs provide some degree of CNS prophylaxis: - Dexamethasone.
- L-asparaginase (does not penetrate into CSF itself, but leads to CSF asparagine depletion).
- High-dose methotrexate with leucovorin rescue.
- Escalating dose intravenous (IV) methotrexate without leucovorin rescue.
Evidence (CNS-directed systemic chemotherapy): - In a randomized Children's Cancer Group (CCG) study of standard-risk patients who all received the same dose and schedule of intrathecal methotrexate without cranial irradiation, oral dexamethasone was associated with a 50% decrease in the rate of CNS relapse compared with oral prednisone.[7]
- In another standard-risk ALL trial (COG-1991), escalating dose IV methotrexate without leucovorin rescue significantly reduced the CNS relapse rate compared with standard, low-dose, oral methotrexate given during each of two interim maintenance phases.[8]
- In a randomized clinical trial conducted by the former Pediatric Oncology Group, T-cell ALL patients who received high-dose methotrexate experienced a significantly lower CNS relapse rate than patients who did not receive high-dose methotrexate.[9]
Cranial Radiation The proportion of patients receiving cranial radiation has decreased significantly over time. At present, most newly diagnosed children with ALL are treated without cranial radiation. Many groups administer cranial radiation only to those patients considered to be at highest risk of subsequent CNS relapse, such as those with documented CNS leukemia at diagnosis (as defined above) (≥5 WBC/μL with blasts; CNS3) and/or T-cell phenotype with high presenting WBC count.[10] In patients who do receive radiation, the cranial radiation dose has been significantly reduced and administration of spinal irradiation is not standard. Ongoing trials seek to determine whether radiation can be eliminated from the treatment of all children with ALL without compromising survival or leading to increased rate of toxicities from upfront and salvage therapies.[11,12] A meta-analysis of randomized trials of CNS-directed therapy has confirmed that radiation therapy can be replaced by intrathecal chemotherapy in most patients with ALL. Additional systemic therapy may be required depending on the agents and intensity used.[13]; [1][Level of evidence: 1iDi] CNS Therapy for Standard-risk Patients Intrathecal chemotherapy without cranial radiation, given in the context of appropriate systemic chemotherapy, results in CNS relapse rates of less than 5% for children with standard-risk ALL.[11,12,14,15,16,17] The use of cranial radiation is not a necessary component of CNS-directed therapy for these patients.[18,19] Some regimens use triple intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone), while others use intrathecal methotrexate alone throughout therapy. Evidence (triple intrathecal chemotherapy vs. intrathecal methotrexate): - The CCG-1952 study for National Cancer Institute (NCI) standard-risk patients compared the relative efficacy and toxicity of triple intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) with methotrexate as the sole intrathecal agent in nonirradiated patients.[20]
- There was no significant difference in either CNS or non-CNS toxicities.
- Although triple intrathecal chemotherapy was associated with a lower rate of isolated CNS relapse (3.4% ± 1.0% compared with 5.9% ± 1.2% for intrathecal methotrexate; P = .004), there was no difference in event-free survival (EFS).
- The reduction in CNS relapse rate was especially notable in patients with CNS2 status at diagnosis (lymphoblasts seen in CSF cytospin, but with <5 WBC/high-power field [hpf] on CSF cell count); the isolated CNS relapse rate was 7.7% ± 5.3% for CNS2 patients who received triple intrathecal chemotherapy compared with 23.0% ± 9.5% for those who received intrathecal methotrexate alone (P = .04).
- There were more bone marrow relapses in the group that received the triple intrathecal chemotherapy, leading to a worse overall survival (OS) (90.3% ± 1.5%) compared with the intrathecal methotrexate group (94.4% ± 1.1%; P = .01).
- When the analysis was restricted to patients with precursor B-cell ALL and rapid early response (M1 marrow on day 14), there was no difference between triple and single intrathecal chemotherapy in terms of rates of CNS relapse rate, OS, or EFS.
- The findings of this trial need to be interpreted within the context of other therapy administered to patients. Dexamethasone, which has been associated with lower CNS relapse rates and improved EFS in standard-risk patients in other trials,[7,21] was not used in CCG-1952 (prednisone was the only steroid administered to patients).[22] It is not clear whether the results of the CCG-1952 trial are generalizable to protocols that include the use of dexamethasone and/or other CNS-directed systemic therapies.
- In a follow-up study of neurocognitive functioning in the two groups, there were no clinically significant differences.[23][Level of evidence: 1iiC]
CNS Therapy for High-risk and Very High-risk Patients Controversy exists as to whether high-risk and very high-risk patients should be treated with cranial radiation. Depending on the protocol, up to 20% of children with ALL receive cranial radiation as part of their CNS-directed therapy, even if they present without CNS involvement at diagnosis. Patients receiving cranial radiation on many treatment regimens include the following:[10] - Patients with T-cell phenotype and high initial WBC count.
- Patients with high-risk precursor B-cell ALL (e.g., extremely high presenting leukocyte counts and/or adverse cytogenetic abnormalities and/or CNS3 disease).
Both the proportion of patients receiving radiation and the dose of radiation administered has decreased over the last 2 decades. Evidence (cranial radiation): - In a trial conducted between 1990 and 1995, the Berlin-Frankfurt-Münster (BFM) group demonstrated that a reduced dose of prophylactic radiation (12 Gy instead of 18 Gy) provided effective CNS prophylaxis in high-risk patients.[24]
- In the follow-up trial conducted by the BFM group between 1995 and 2000 (BFM-95), cranial radiation was administered to approximately 20% of patients (compared with 70% on the previous trial), including patients with T-cell phenotype, a slow early response (as measured by peripheral blood blast count after a 1-week steroid prophase), and/or adverse cytogenetic abnormalities.[17]
- While the rate of isolated CNS relapses was higher in the nonirradiated higher-risk patients compared with historic (irradiated) cohorts, their overall EFS rate was not significantly different.
- Several groups, including the St. Jude Children's Research Hospital (SJCRH), the Dutch Childhood Oncology Group (DCOG), and the European Organization for Research and Treatment of Cancer (EORTC), have published results of trials that omitted cranial radiation for all patients, including high-risk subsets.[11,12,25] Most of these trials have included at least four doses of high-dose methotrexate during postinduction consolidation and an increased frequency of intrathecal chemotherapy. The SJCRH and DCOG studies also included frequent vincristine/dexamethasone pulses and intensified dosing of pegaspargase,[11,12] while the EORTC trials included additional high-dose methotrexate and multiple doses of high-dose cytarabine during postinduction treatment phases for CNS3 (CSF with ≥5 WBC/µL and cytospin positive for blasts) patients.[25]
- The 5-year cumulative incidence of isolated CNS relapse on those trials was between 2% and 4%, although some patient subsets had a significantly higher rate of CNS relapse. On the SJCRH study, clinical features associated with a significantly higher risk of isolated CNS relapse included T-cell phenotype, the t(1;19) translocation, or the presence of blasts in the CSF at diagnosis.[11]
- The overall EFS for the SJCRH study was 85.6% and 81% for the DCOG study, both in line with outcomes achieved by contemporaneously conducted clinical trials on which some patients received prophylactic radiation, but was lower on the EORTC trial (8-year EFS, 69.6%).[25]
- Of note, on the SJCRH study, 33 of 498 (6.6%) patients in first remission with high-risk features (including 26 with high minimal residual disease (MRD), six with Philadelphia chromosome-positive ALL, and one with near haploidy) received an allogeneic hematopoietic stem cell transplant , which included total-body irradiation.[11]
- In a meta-analysis of aggregated data from more than 16,000 patients treated between 1996 and 2007 by ten cooperative groups, the use of cranial radiation therapy did not appear to impact 5-year OS or cumulative incidence of any event.[13]
- In subgroup analyses of high-risk subsets, only those with CNS3 status at diagnosis appeared to benefit from cranial radiation, with a significantly lower rate of CNS relapses (isolated/any) in irradiated patients; however, even within this subgroup, OS was similar with or without the use of radiation therapy.
- This study suggests that cranial radiation therapy may not be an essential component of treatment, even for high-risk patients; however, interpretation is limited by the considerable variation in treatment administered to patients by the different cooperative groups.
CNS Therapy for Patients With CNS Involvement (CNS3 Disease) at Diagnosis Therapy for ALL patients with clinically evident CNS disease (≥5 WBC/hpf with blasts on cytospin; CNS3) at diagnosis typically includes intrathecal chemotherapy and cranial radiation (usual dose is 18 Gy).[17,19] Spinal radiation is no longer used. Evidence (cranial radiation): - SJCRH, DCOG, and the EORTC have published results of trials that omitted cranial radiation for all patients, including high-risk subsets.[11,25] These trials have included at least four doses of high-dose methotrexate during postinduction consolidation and an increased frequency of intrathecal chemotherapy. The SJCRH study also included higher cumulative doses of anthracycline than on Children's Oncology Group (COG) trials, and frequent vincristine/dexamethasone pulses and intensified dosing of pegaspargase,[11] while the EORTC trials included additional high-dose methotrexate and multiple doses of high-dose cytarabine, during postinduction treatment phases for CNS3 (CSF with ≥5 WBC/µL and cytospin positive for blasts) patients.[25]
- On the SJCRH Total XV (TOTXV) study, patients with CNS3 status (N = 9) were treated without cranial radiation (observed 5-year EFS, 43% ± 23%%; OS, 71% ± 22%).[11] On this study, CNS leukemia at diagnosis (defined as CNS3 status or traumatic lumbar puncture with blasts) was an independent predictor of inferior EFS.
- On the DCOG-9 trial, the 5-year EFS of CNS3 patients (n = 21) treated without cranial radiation was 67% ± 10%.[12]
- On the EORTC trial, the 8-year EFS of CNS3 patients (n = 49) treated without cranial radiation was 68%. The cumulative incidence of isolated CNS relapse for those patients was 9.4%.[25][Level of evidence: 2A]
- A meta-analysis of aggregated data from more than 16,000 patients treated between 1996 and 2007 by ten cooperative groups evaluated whether the use of cranial radiation therapy affected outcome in high-risk patient subsets.[13]
- In subgroup analyses of high-risk subsets, only those with CNS3 status at diagnosis appeared to benefit from cranial radiation therapy, with a significantly lower rate of CNS relapses (isolated/any) in irradiated patients; however, even within this subgroup, OS was similar with or without the use of radiation therapy.
Larger prospective studies will be necessary to fully elucidate the safety of omitting cranial radiation in CNS3 patients. Presymptomatic CNS Therapy Options Under Clinical Evaluation Treatment options under clinical evaluation include the following: - NCI-2014-00712; AALL1231 (NCT02112916) (Combination Chemotherapy With or Without Bortezomib in Treating Younger Patients With Newly Diagnosed T-Cell ALL or Stage II-IV T-Cell Lymphoblastic Lymphoma): This trial is for patients with T-cell ALL and is testing, in a nonrandomized fashion, reduction in the proportion of T-cell ALL patients who receive prophylactic cranial radiation. In this study, only very high-risk patients (those with M3 marrow at day 29 or MRD >0.1% at end of consolidation, regardless of initial CNS status) and any other patient who is CNS3 at diagnosis receive cranial radiation therapy. CNS3 patients receive 18 Gy of cranial radiation, while the other patients allocated to cranial radiation receive 12 Gy. It is estimated that 10% to 15% of T-cell ALL patients will receive cranial radiation on AALL1231, compared with 85% to 90% of T-cell ALL patients on predecessor COG trials.
- COG-AALL1131 (NCT01406756) (Combination Chemotherapy in Treating Young Patients With Newly Diagnosed High-Risk ALL): The COG-AALL1131 protocol for patients with high-risk precursor B-cell ALL includes a randomized comparison of triple intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) with intrathecal methotrexate, with the objective of determining whether triple intrathecal chemotherapy reduces CNS-relapse rates and improves overall EFS. Only patients with CNS3 status at diagnosis will receive cranial radiation (18 Gy).
- SJCRH Total XVI (TOTXVI; NCT00549848) (Total Therapy Study XVI for Newly Diagnosed Patients With ALL): Patients receive both intrathecal chemotherapy and high-dose methotrexate without radiation therapy. Certain patients with high-risk features, including those with a t(1;19) translocation, receive intensified intrathecal therapy.
Toxicity of CNS-Directed Therapy Toxic effects of CNS-directed therapy for childhood ALL can be acute and subacute or late developing. (Refer to the Late Effects of the Central Nervous System section in the PDQ summary on Late Effects of Treatment for Childhood Cancer for more information.) Acute and subacute toxicities The most common acute side effect associated with intrathecal chemotherapy alone is seizures. Up to 5% of nonirradiated patients with ALL treated with frequent doses of intrathecal chemotherapy will have at least one seizure during therapy.[11] Higher rates of seizure were observed with consolidation regimens that included multiple doses of high-dose methotrexate in addition to intrathecal chemotherapy.[26] Intrathecal and high-dose intravenous methotrexate has also been associated with a stroke-like syndrome, which, in most cases, appears to be reversible.[27] Patients with ALL who develop seizures during the course of treatment and who receive anticonvulsant therapy should not receive phenobarbital or phenytoin as anticonvulsant treatment, as these drugs may increase the clearance of some chemotherapeutic drugs and adversely affect treatment outcome.[28] Gabapentin or valproic acid are alternative anticonvulsants with less enzyme-inducing capabilities.[28] Late-developing toxicities Late effects associated with CNS-directed therapies include subsequent neoplasms, neuroendocrine disturbances, leukoencephalopathy, and neurocognitive impairments. Subsequent neoplasms are observed primarily in survivors who received cranial radiation. Meningiomas are common and typically of low malignant potential, but high-grade lesions also occur. In a SJCRH retrospective study of more than 1,290 ALL patients who had never relapsed, the 30-year cumulative incidence of a subsequent neoplasm occurring in the CNS was 3%; excluding meningiomas, the 30-year cumulative incidence was 1.17%.[29] Nearly all of these CNS subsequent neoplasms occurred in previously irradiated patients. Neurocognitive impairments, which can range in severity and functional consequences, have been documented in long-term ALL survivors treated both with and without radiation therapy. In general, patients treated without cranial radiation have less severe neurocognitive sequelae than irradiated patients, and the deficits that do develop represent relatively modest declines in a limited number of domains of neuropsychological functioning.[30,31,32,33] For patients who receive cranial radiation therapy, the frequency and severity of toxicities appear dose-related; patients treated with 18 Gy of cranial radiation therapy appear to be at lower risk for severe impairments compared with those treated with doses of 24 Gy or higher. Younger age at diagnosis and female sex have been reported in many studies to be associated with a higher risk of neurocognitive late effects.[34] Several studies have also evaluated the impact of other components of treatment on the development of late neurocognitive impairments. A comparison of neurocognitive outcomes of patients treated with methotrexate versus triple intrathecal chemotherapy showed no clinically meaningful difference.[23][Level of evidence: 3iiiC] Controversy exists about whether patients who receive dexamethasone have a higher risk of neurocognitive disturbances.[35] In a SJCRH study of nonirradiated long-term survivors, treatment with dexamethasone was associated with increased risk for impairments in attention and executive function.[36] Conversely, long-term neurocognitive testing in 92 children with a history of standard-risk ALL who had received either dexamethasone or prednisone during treatment did not demonstrate any meaningful differences in cognitive functioning based on corticosteroid randomization.[37] Evidence (neurocognitive late effects of cranial radiation): - A SJCRH study of 567 adult long-term survivors of childhood ALL underwent neurocognitive testing (mean time from diagnosis, 26 years).[36]
- Patients treated with 24 Gy of cranial radiation showed the highest rates of impairment. Up to one-third of these patients demonstrated impairments (defined as test scores 2 or more standard deviations below age-adjusted national norms) in attention, memory, processing speed, and executive function.
- Significantly fewer patients who had received 18 Gy of cranial radiation demonstrated severe impairments compared with those who had received 24 Gy. In general, there was no significant difference in rates of impairment between nonirradiated survivors and those who received 18 Gy of cranial radiation; however, the 18 Gy group was at increased risk for academic problems.
- In addition to being dose-related, the neurocognitive impact of cranial radiation was also dependent on age at diagnosis, with higher frequency of impairments in patients diagnosed at a younger age.
- A study compared memory impairment in patients receiving 18 Gy of cranial radiation (n = 127) versus 24 Gy of cranial radiation (n = 138).[38]
- Long-term survivors who received 24 Gy, but not 18 Gy, of cranial radiation demonstrated significant impairments in immediate and delayed memory.
- In a randomized trial comparing irradiated (at a dose of 18 Gy) and nonirradiated standard-risk ALL patients, the following was observed: [30][Level of evidence: 1iiC]
- Cognitive function for both groups (assessed at a median of 6 years postdiagnosis) was in the average range, with only subtle differences noted between the groups in cognitive skills.
- In a randomized trial, hyperfractionated radiation (at a dose of 18 Gy) did not decrease neurologic late effects when compared with conventionally fractionated radiation; cognitive function for both groups was not significantly impaired.[39]
Evidence (neurocognitive late effects in nonirradiated patients): - In the SJCRH long-term follow-up study of 567 adult long-term survivors, some nonirradiated patients also demonstrated neurocognitive impairments.[36]
- The age-adjusted mean test scores for nonirradiated patients were very similar to that of expected national norms; however, approximately 15% of the nonirradiated survivors participating in this study demonstrated impairments in some domains, including attention, memory, processing speed, and executive function.
- Despite the impairments noted on neurocognitive testing, overall, the educational attainment and employment status of the tested ALL survivors were similar to age- and sex-adjusted expected proportions using census data for the U.S. population.
- In a second study from SJCRH, patients enrolled on Total Study XV (which omitted cranial radiation in all patients) underwent comprehensive neuropsychological assessments at induction, end of maintenance, and 2 years after completion of therapy.[40]
- Neurocognitive function was largely age appropriate 2 years after completing therapy, without evidence of excess impairment on measures of intellectual functioning, academic abilities, learning, and memory. Problems with sustained attention were observed at an increased frequency in this population compared with normative expectations.
- High-risk patients who received more intensive CNS-directed chemotherapy (including high-dose methotrexate and more doses of intrathecal chemotherapy) were at greater risk for difficulties in attention, processing speed, and academics.
References:
-
Richards S, Pui CH, Gayon P, et al.: Systematic review and meta-analysis of randomized trials of central nervous system directed therapy for childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 60 (2): 185-95, 2013.
-
Mahmoud HH, Rivera GK, Hancock ML, et al.: Low leukocyte counts with blast cells in cerebrospinal fluid of children with newly diagnosed acute lymphoblastic leukemia. N Engl J Med 329 (5): 314-9, 1993.
-
Bürger B, Zimmermann M, Mann G, et al.: Diagnostic cerebrospinal fluid examination in children with acute lymphoblastic leukemia: significance of low leukocyte counts with blasts or traumatic lumbar puncture. J Clin Oncol 21 (2): 184-8, 2003.
-
Gajjar A, Harrison PL, Sandlund JT, et al.: Traumatic lumbar puncture at diagnosis adversely affects outcome in childhood acute lymphoblastic leukemia. Blood 96 (10): 3381-4, 2000.
-
Pullen J, Boyett J, Shuster J, et al.: Extended triple intrathecal chemotherapy trial for prevention of CNS relapse in good-risk and poor-risk patients with B-progenitor acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 11 (5): 839-49, 1993.
-
Thyss A, Suciu S, Bertrand Y, et al.: Systemic effect of intrathecal methotrexate during the initial phase of treatment of childhood acute lymphoblastic leukemia. The European Organization for Research and Treatment of Cancer Children's Leukemia Cooperative Group. J Clin Oncol 15 (5): 1824-30, 1997.
-
Bostrom BC, Sensel MR, Sather HN, et al.: Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: a report from the Children's Cancer Group. Blood 101 (10): 3809-17, 2003.
-
Matloub Y, Bostrom BC, Hunger SP, et al.: Escalating intravenous methotrexate improves event-free survival in children with standard-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 (2): 243-51, 2011.
-
Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.
-
Pui CH, Howard SC: Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol 9 (3): 257-68, 2008.
-
Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
-
Veerman AJ, Kamps WA, van den Berg H, et al.: Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 10 (10): 957-66, 2009.
-
Vora A, Andreano A, Pui CH, et al.: Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 34 (9): 919-26, 2016.
-
Pui CH, Sandlund JT, Pei D, et al.: Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood 104 (9): 2690-6, 2004.
-
Tubergen DG, Gilchrist GS, O'Brien RT, et al.: Prevention of CNS disease in intermediate-risk acute lymphoblastic leukemia: comparison of cranial radiation and intrathecal methotrexate and the importance of systemic therapy: a Childrens Cancer Group report. J Clin Oncol 11 (3): 520-6, 1993.
-
Conter V, Aricò M, Valsecchi MG, et al.: Extended intrathecal methotrexate may replace cranial irradiation for prevention of CNS relapse in children with intermediate-risk acute lymphoblastic leukemia treated with Berlin-Frankfurt-Münster-based intensive chemotherapy. The Associazione Italiana di Ematologia ed Oncologia Pediatrica. J Clin Oncol 13 (10): 2497-502, 1995.
-
Möricke A, Reiter A, Zimmermann M, et al.: Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111 (9): 4477-89, 2008.
-
Clarke M, Gaynon P, Hann I, et al.: CNS-directed therapy for childhood acute lymphoblastic leukemia: Childhood ALL Collaborative Group overview of 43 randomized trials. J Clin Oncol 21 (9): 1798-809, 2003.
-
Moghrabi A, Levy DE, Asselin B, et al.: Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 109 (3): 896-904, 2007.
-
Matloub Y, Lindemulder S, Gaynon PS, et al.: Intrathecal triple therapy decreases central nervous system relapse but fails to improve event-free survival when compared with intrathecal methotrexate: results of the Children's Cancer Group (CCG) 1952 study for standard-risk acute lymphoblastic leukemia, reported by the Children's Oncology Group. Blood 108 (4): 1165-73, 2006.
-
Mitchell CD, Richards SM, Kinsey SE, et al.: Benefit of dexamethasone compared with prednisolone for childhood acute lymphoblastic leukaemia: results of the UK Medical Research Council ALL97 randomized trial. Br J Haematol 129 (6): 734-45, 2005.
-
Vrooman LM, Stevenson KE, Supko JG, et al.: Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol 31 (9): 1202-10, 2013.
-
Kadan-Lottick NS, Brouwers P, Breiger D, et al.: Comparison of neurocognitive functioning in children previously randomly assigned to intrathecal methotrexate compared with triple intrathecal therapy for the treatment of childhood acute lymphoblastic leukemia. J Clin Oncol 27 (35): 5986-92, 2009.
-
Schrappe M, Reiter A, Ludwig WD, et al.: Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95 (11): 3310-22, 2000.
-
Sirvent N, Suciu S, Rialland X, et al.: Prognostic significance of the initial cerebro-spinal fluid (CSF) involvement of children with acute lymphoblastic leukaemia (ALL) treated without cranial irradiation: results of European Organization for Research and Treatment of Cancer (EORTC) Children Leukemia Group study 58881. Eur J Cancer 47 (2): 239-47, 2011.
-
Mahoney DH Jr, Shuster JJ, Nitschke R, et al.: Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy--a Pediatric Oncology Group study. J Clin Oncol 16 (5): 1712-22, 1998.
-
Bhojwani D, Sabin ND, Pei D, et al.: Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J Clin Oncol 32 (9): 949-59, 2014.
-
Relling MV, Pui CH, Sandlund JT, et al.: Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet 356 (9226): 285-90, 2000.
-
Hijiya N, Hudson MM, Lensing S, et al.: Cumulative incidence of secondary neoplasms as a first event after childhood acute lymphoblastic leukemia. JAMA 297 (11): 1207-15, 2007.
-
Waber DP, Turek J, Catania L, et al.: Neuropsychological outcomes from a randomized trial of triple intrathecal chemotherapy compared with 18 Gy cranial radiation as CNS treatment in acute lymphoblastic leukemia: findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J Clin Oncol 25 (31): 4914-21, 2007.
-
Jansen NC, Kingma A, Schuitema A, et al.: Neuropsychological outcome in chemotherapy-only-treated children with acute lymphoblastic leukemia. J Clin Oncol 26 (18): 3025-30, 2008.
-
Espy KA, Moore IM, Kaufmann PM, et al.: Chemotherapeutic CNS prophylaxis and neuropsychologic change in children with acute lymphoblastic leukemia: a prospective study. J Pediatr Psychol 26 (1): 1-9, 2001 Jan-Feb.
-
Copeland DR, Moore BD 3rd, Francis DJ, et al.: Neuropsychologic effects of chemotherapy on children with cancer: a longitudinal study. J Clin Oncol 14 (10): 2826-35, 1996.
-
von der Weid N, Mosimann I, Hirt A, et al.: Intellectual outcome in children and adolescents with acute lymphoblastic leukaemia treated with chemotherapy alone: age- and sex-related differences. Eur J Cancer 39 (3): 359-65, 2003.
-
Waber DP, Carpentieri SC, Klar N, et al.: Cognitive sequelae in children treated for acute lymphoblastic leukemia with dexamethasone or prednisone. J Pediatr Hematol Oncol 22 (3): 206-13, 2000 May-Jun.
-
Krull KR, Brinkman TM, Li C, et al.: Neurocognitive outcomes decades after treatment for childhood acute lymphoblastic leukemia: a report from the St Jude lifetime cohort study. J Clin Oncol 31 (35): 4407-15, 2013.
-
Kadan-Lottick NS, Brouwers P, Breiger D, et al.: A comparison of neurocognitive functioning in children previously randomized to dexamethasone or prednisone in the treatment of childhood acute lymphoblastic leukemia. Blood 114 (9): 1746-52, 2009.
-
Armstrong GT, Reddick WE, Petersen RC, et al.: Evaluation of memory impairment in aging adult survivors of childhood acute lymphoblastic leukemia treated with cranial radiotherapy. J Natl Cancer Inst 105 (12): 899-907, 2013.
-
Waber DP, Silverman LB, Catania L, et al.: Outcomes of a randomized trial of hyperfractionated cranial radiation therapy for treatment of high-risk acute lymphoblastic leukemia: therapeutic efficacy and neurotoxicity. J Clin Oncol 22 (13): 2701-7, 2004.
-
Jacola LM, Krull KR, Pui CH, et al.: Longitudinal Assessment of Neurocognitive Outcomes in Survivors of Childhood Acute Lymphoblastic Leukemia Treated on a Contemporary Chemotherapy Protocol. J Clin Oncol 34 (11): 1239-47, 2016.
Postinduction Treatment for Specific ALL SubgroupsT-Cell ALL Historically, patients with T-cell acute lymphoblastic leukemia (ALL) have had a worse prognosis than children with precursor B-cell ALL. In a review of a large number of patients treated on Children's Oncology Group (COG) trials over a 15-year period, T-cell immunophenotype still proved to be a negative prognostic factor on multivariate analysis.[1] However, with current treatment regimens, outcomes for children with T-cell ALL are now approaching those achieved for children with precursor B-cell ALL. For example, the 10-year overall survival (OS) for children with T-cell ALL treated on the Dana-Farber Cancer Institute (DFCI) DFCI-95001 (NCT00004034) trial was 90.1%, compared with 88.7% for patients with B-cell disease.[2] Another example is the COG trial for T-cell ALL (AALL0434 [NCT00408005]) that observed a 4-year disease-free survival (DFS) of 89.3%.[3] Treatment options for T-cell ALL - Protocols of the former Pediatric Oncology Group (POG) treated children with T-cell ALL differently from children with B-lineage ALL. The POG-9404 protocol for patients with T-cell ALL was designed to evaluate the role of high-dose methotrexate. The multiagent chemotherapy regimen for this protocol was based on the DFCI-87001 regimen.[4]
- Results of the POG-9404 study indicated that the addition of high-dose methotrexate to the DFCI-based chemotherapy regimen resulted in significantly improved event-free survival (EFS) in patients with T-cell ALL (10-year EFS, 78% for those randomly assigned to high-dose methotrexate versus 68% for those randomly assigned to therapy without high-dose methotrexate, P = .05).
- High-dose methotrexate was associated with a lower incidence of relapses involving the central nervous system (CNS).[5] This POG study was the first clinical trial to provide evidence that high-dose methotrexate can improve outcome for children with T-cell ALL. High-dose asparaginase, doxorubicin, and prophylactic cranial irradiation were also important components of this regimen.[2,5]
- In the POG-9404 study, patients were randomly assigned to doxorubicin with or without dexrazoxane to determine the efficacy of dexrazoxane in preventing late cardiac mortality.[6][Level of evidence: 1iiDi]
- There was no difference in EFS between patients with T-cell ALL who were treated with dexrazoxane and patients who were not treated with dexrazoxane (cumulative doxorubicin dose, 360 mg/m2).[6]
- The frequency of grade 3 and grade 4 toxicities that occurred during therapy was similar between the randomized groups, and there was no difference in cumulative incidence of second malignant neoplasms. Three years after initial diagnosis, left ventricular shortening fraction and left ventricular wall thickness were both significantly worse in patients who received doxorubicin alone than in patients who received dexrazoxane, indicating that dexrazoxane was cardioprotective.[6]
- With combined data from three COG trials that randomized dexrazoxane with doxorubicin therapy (P9404, P9425, and P9426) and had a median follow-up of 12.6 years, dexrazoxane did not appear to compromise long-term survival.[7][Level of evidence: 1iiA]
- Protocols of the former Children's Cancer Group (CCG) treated children with T-cell ALL on the same treatment regimens as children with precursor B-cell ALL, basing protocol and treatment assignment on the patients' clinical characteristics (e.g., age and white blood cell [WBC] count) and the disease response to initial therapy. Most children with T-cell ALL meet National Cancer Institute (NCI) high-risk criteria.
- Results from CCG-1961 for high-risk ALL including T-cell ALL showed that an augmented Berlin-Frankfurt-Münster (BFM) regimen with a single delayed intensification course produced the best results for patients with morphologic rapid response to initial induction therapy (estimated 5-year EFS, 83%).[8,9] With this approach, patients with a presenting WBC count greater than 200,000 had similar outcomes to those with a WBC count of less than 200,000.[10][Level of evidence: 1iiDi]
- Overall results from POG-9404 and CCG-1961 were similar, although POG-9404 used higher cumulative dose of anthracyclines and cranial radiation for every patient, while CCG-1961 used cranial radiation only for patients with slow morphologic response.[9,5]
- Among children with NCI standard-risk T-cell ALL, the 7-year EFS for those treated on CCG-1952, COG-1991 and POG-9404 is comparable with the CCG regimens utilizing significantly less anthracycline in a less intensive chemotherapy backbone without the prophylactic cranial irradiation included in POG-9404.[11]
- In the COG, children with T-cell ALL are not treated on the same protocols as children with precursor B-cell ALL.
- Pilot studies from the COG have demonstrated the feasibility of incorporating nelarabine (a nucleoside analog with demonstrated activity in patients with relapsed and refractory T-cell lymphoblastic disease) [12,13,14] in the context of a BFM regimen for patients with newly diagnosed T-cell ALL. The pilot study showed a 5-year EFS rate of 73% for all patients receiving nelarabine and 69% for those patients with a slow early response.[15]
- The COG AALL0434 (NCT00408005) trial treated patients with T-cell ALL on an augmented BFM regimen and randomly assigned patients to receive either high-dose methotrexate with leucovorin rescue or escalating methotrexate without leucovorin (Capizzi).[3] All patients received either prophylactic (12 Gy) or therapeutic (18 Gy) cranial irradiation except for low-risk patients. Results indicate a better EFS for patients randomly assigned to the Capizzi arm: 92.5% 4-year EFS vs. 86.1% for high-dose methotrexate (P = .0173).[16]
- The use of prophylactic cranial radiation in the treatment of T-cell ALL is declining. Some groups, such as St. Jude Children's Research Hospital (SJCRH) and the Dutch Childhood Oncology Group (DCOG), do not use cranial radiation in first-line treatment of ALL, and other groups, such as DFCI, COG, and BFM, are now limiting radiation to patients with very high-risk features or CNS3 disease.
Treatment options under clinical evaluation for T-cell ALL Treatment options under clinical evaluation for T-cell ALL include the following: - NCI-2014-00712; AALL1231 (NCT02112916)(Combination Chemotherapy With or Without Bortezomib in Treating Younger Patients With Newly Diagnosed T-Cell ALL or Stage II-IV T-Cell Lymphoblastic Lymphoma): This phase III trial is utilizing a modified augmented BFM regimen for patients aged 1 to 30 years with T-cell ALL. Patients are classified into one of three risk groups (standard, intermediate, or very high) based on morphologic response at day 29, minimal residual disease (MRD) status at day 29 and end of consolidation, and CNS status at diagnosis. Age and presenting leukocyte count are not used to stratify patients. The objectives of the trial include the following:
- To compare EFS in patients who are randomly assigned to receive or not to receive bortezomib on a modified augmented BFM backbone. For those randomly assigned to receive bortezomib, it is given during the induction phase (four doses) and again during the delayed intensification phase (four doses).
- To determine the safety and feasibility of modifying standard COG therapy for T-cell ALL by using dexamethasone instead of prednisone during the induction and maintenance phases and additional doses of pegaspargase during the induction and delayed intensification phases.
- To determine whether prophylactic cranial radiation can be omitted in 85% to 90% of T-cell ALL patients (non-very high risk, non-CNS3) without an increase in relapse risk, compared with historic controls.
- To determine the proportion of patients with end consolidation MRD >0.1% who become MRD-negative after intensification therapy using three high-risk BFM blocks that include high-dose cytarabine, high-dose methotrexate, ifosfamide, and etoposide.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with T-cell childhood acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. Infants With ALL Infant ALL is uncommon, representing approximately 2% to 4% of cases of childhood ALL.[17] Because of their distinctive biological characteristics and their high risk of leukemia recurrence, infants with ALL are treated on protocols specifically designed for this patient population. Common therapeutic themes of the intensive chemotherapy regimens used to treat infants with ALL are the inclusion of postinduction intensification courses with high doses of cytarabine and methotrexate.[18,19,20] Infants diagnosed within the first few months of life have a particularly poor outcome. In one study, patients diagnosed within 1 month of birth had a 2-year OS rate of 20%.[21][Level of evidence: 2A] In another study, the 5-year EFS for infants diagnosed at younger than 90 days was 16%.[20][Level of evidence: 2A] For infants with MLL (KMT2A) gene rearrangement, the EFS rates continue to be in the 35% range.[18,19,20,22][Level of evidence: 2A] Factors predicting poor outcome for infants with MLL rearrangements include the following:[19,20]; [23][Level of evidence: 3iDii] - A very young age (≤90 days).
- Extremely high presenting leukocyte count (≥200,000-300,000/μL).
- Poor early response, as reflected by a poor response to a prednisone prophase or high levels of MRD at the end of induction and consolidation phases of treatment.
Infants have significantly higher relapse rates than older children with ALL and are at higher risk of developing treatment-related toxicity, especially infection. With current treatment approaches for this population, treatment-related mortality has been reported to occur in about 10% of infants, a rate that is much higher than the rate in older children with ALL.[19,20] On the COG AALL0631 (NCT00557193) trial, an intensified induction regimen resulted in an induction death rate of 15.4% (4 of 26 patients); the trial was subsequently amended to include a less-intensive induction and enhanced supportive care guidelines, resulting in a significantly lower induction death rate (1.6%; 2 of 123 patients) and significantly higher complete remission (CR) rate (94% vs. 68% with the previous, more intensified induction regimen).[24] Treatment options for infants withMLL(KMT2A) rearrangements Infants with MLL (KMT2A) gene rearrangements are generally treated on intensified chemotherapy regimens using agents not typically incorporated into frontline therapy for older children with ALL. However, despite these intensified approaches, EFS rates remain poor for these patients. Evidence (intensified chemotherapy regimens for infants with MLL [KMT2A] rearrangements): - The international Interfant clinical trials consortium utilized a cytarabine-intensive chemotherapy regimen, with increased exposure to both low- and high-dose cytarabine during the first few months of therapy, resulting in a 5-year EFS of 37% for infants with MLL (KMT2A) rearrangements.[19]
- The COG tested intensification of therapy with a regimen including multiple doses of high-dose methotrexate, cyclophosphamide, and etoposide, resulting in a 5-year EFS of 34% for infants with MLL rearrangements.[18]
- On the COG P9407 (NCT00002756) trial, infants were treated with a shortened (46-week) intensive chemotherapy regimen. The 5-year EFS for infants with MLL rearrangements was 36%.[20][Level of evidence: 2A]
The role of allogeneic hematopoietic stem cell transplant (HSCT) during first remission in infants with MLL (KMT2A) gene rearrangements remains controversial. Evidence (allogeneic HSCT in first remission for infants with MLL [KMT2A] rearrangements): - On a Japanese clinical trial conducted between 1998 and 2002, all infants with MLL (KMT2A)-rearrangement were intended to proceed to allogeneic HSCT from the best available donor (related, unrelated, or umbilical cord) 3 to 5 months after diagnosis.[25]
- The 3-year EFS for all enrolled infants was 44%. This result was due, in part, to the high frequency of early relapses, even with intensive chemotherapy; of the 41 infants with MLL rearrangement on that study who achieved CR, 11 infants (27%) relapsed before proceeding to transplant.
- In a COG report that included 189 infants treated on CCG or POG infant ALL protocols between 1996 and 2000, there was no difference in EFS between patients who underwent HSCT in first CR and those who received chemotherapy alone.[26]
- The Interfant clinical trials group, after adjusting for waiting time to transplantation, also did not observe any difference in DFS in high-risk infants (defined by prednisone response) with MLL (KMT2A) rearrangements treated on the Interfant-99 trial with either allogeneic HSCT in first CR or chemotherapy alone.[19]
- In a subset analysis from the same trial, allogeneic HSCT in first remission was associated with a significantly better DFS for infants with MLL rearrangements who were younger than 6 months at diagnosis and had either a poor prednisone response at day 8 or leukocyte counts of at least 300,000/µL.[27] In this subset, HSCT in first remission was associated with a 64% reduction in the risk of failure resulting from relapse or death compared with chemotherapy alone.
- For infants with ALL who undergo transplantation in first CR, outcomes appear to be similar with non-total-body irradiation (TBI) regimens and TBI-based regimens.[26,28]
Treatment options for infants withoutMLL(KMT2A) rearrangements The optimal treatment for infants without MLL (KMT2A) rearrangements also remains unclear. - On the Interfant-99 trial, patients without MLL (KMT2A) rearrangement achieved a relatively favorable outcome with the cytarabine-intensive treatment regimen (4-year EFS was 74%).[19]
- The COG P9407 (NCT00002756) trial of intensified chemotherapy reported a 70% 5-year EFS in infants without the MLL rearrangement.[20][Level of evidence: 2A]
- A favorable outcome for this subset of patients was obtained in a Japanese study using therapy comparable to that used to treat older children with ALL;[22] however, that study was limited by small numbers (n = 22) and a highly unusual gender distribution (91% males).
Treatment options under clinical evaluation for infants with ALL Treatment options under clinical evaluation include the following: - Interfant-06 Study Group trial (DCOG-INTERFANT-06) (Different Therapies in Treating Infants With Newly Diagnosed Acute Leukemia): The Interfant-06 Study Group is conducting an international collaborative randomized trial (including sites in the United States) to test whether an ALL/acute myeloid leukemia hybrid regimen might improve outcomes for infants with MLL (KMT2A)-rearranged ALL. The role of allogeneic transplantation in first remission is also being assessed in high-risk patients (defined as infants with MLL-rearranged ALL, younger than 6 months, and WBC >300,000 /µL) or poor peripheral blood response to steroid prophase. Infants with MLL-rearranged ALL with high MRD at end of consolidation phase are also eligible for allogeneic HSCT in first remission regardless of other presenting features.
Adolescents and Young Adults With ALL Adolescents and young adults with ALL have been recognized as high risk for decades. Outcomes in almost all studies of treatment are inferior in this age group compared with children younger than 10 years.[29,30,31] The reasons for this difference include more frequent presentation of adverse prognostic factors at diagnosis, including the following: - T-cell immunophenotype.
- Philadelphia chromosome-positivity (Ph+) and BCR-ABL1-like (Ph-like) disease.
- Lower incidence of favorable cytogenetic abnormalities.
In addition to more frequent adverse prognostic factors, patients in this age group have higher rates of treatment-related mortality [30,31,32,33] and nonadherence to therapy.[32,34] Treatment options Studies from the United States and France were among the first to identify the difference in outcome based on treatment regimens.[35] Other studies have confirmed that older adolescent and young adult patients fare better on pediatric rather than adult regimens.[35,36,37,38,39,40,41,42]; [43][Level of evidence: 2A] These study results are summarized in Table 3. Given the relatively favorable outcome that can be obtained in these patients with chemotherapy regimens used for high-risk pediatric ALL, there is no role for the routine use of allogeneic HSCT for adolescents and young adults with ALL in first remission.[31] Evidence (use of a pediatric treatment regimen for adolescents and young adults with ALL): - Investigators reported on 197 patients aged 16 to 21 years treated on the CCG study (a pediatric ALL regimen) who showed a 7-year EFS of 63% compared with 124 adolescents and young adults treated on the Cancer and Leukemia Group B (CALGB) study (an adult ALL regimen) with a 7-year EFS of 34%.[35]
- A study from France of patients aged 15 to 20 years and diagnosed between 1993 and 1999 demonstrated superior outcome for patients treated on a pediatric trial (67%; 5-year EFS) compared with patients treated on an adult trial (41%; 5-year EFS).[40]
- In the COG high-risk study (CCG-1961), the 5-year EFS rate for 262 patients aged 16 to 21 years was 71.5%.[31][Level of evidence: 1iiDi] For rapid responders randomly assigned to early intensive postinduction therapy on the augmented intensity arms of this study, the 5-year EFS rate was 82% (n = 88).
- The DFCI ALL Consortium reported that a study of 51 adolescents aged 15 to 18 years in a pediatric trial had a 5-year EFS of 78%.[37]
- In an SJCRH study, 44 adolescents aged 15 to 18 years had an EFS of approximately 85% ± 5%.[30]
- In a Spanish study, 35 adolescents (aged 15-18 years) and 46 young adults (aged 19-30 years) with standard-risk ALL were treated with a pediatric-based regimen.[43][Level of evidence: 2A]
- EFS rate was 61%.
- The OS rate was 69%.
- There were no differences in outcome between adolescents and young adults.
- In a trial conducted in Japan, 139 adolescent and young adult patients (aged 15-24 years) with Philadelphia chromosome-negative ALL were treated with a high-risk pediatric regimen.[44]
- The 5-year DFS rate was 67%, and the 5-year OS rate was 73%-significantly better than the outcome observed in similar-aged patients treated in the predecessor trial using a nonpediatric regimen (5-year DFS, 44% and OS, 45%).
- There did not appear to be more severe adverse events in adolescent and young adult patients than in younger patients treated with the same high-risk regimen.
- Twenty-one percent of the adolescent and young adult patients did not receive the full intended course of treatment; these patients had a significantly worse DFS.
- The UKALL2003 (NCT00222612) trial studied treatment intensification according to MRD at the end of induction. Patients aged 16 to 24 years (N = 229) who were Philadelphia chromosome negative were enrolled in the study.[45][Level of evidence: 1iiDi]
- Five-year EFS overall for this group was 72%.
- Five-year EFS for low risk patients was 93%.
- There were more serious adverse events in this age group than in younger patients.
Other studies have confirmed that older adolescent patients and young adults fare better on pediatric rather than adult regimens (refer to Table 3).[36,38,41,42] ; [43][Level of evidence: 2A] The reason that adolescents and young adults achieve superior outcomes with pediatric regimens is not known, although possible explanations include the following: [36] - Treatment setting (i.e., site experience in treating ALL).
- Adherence to protocol therapy.
- The components of protocol therapy.
Table 3. Outcome According to Treatment Protocol for Adolescents and Young Adults with ALL| Site and Study Group | Adolescent and Young Adult Patients (No.) | Median age (y) | Survival (%) |
|---|
| ALL = acute lymphoblastic leukemia; EFS = event-free survival; OS = overall survival. | | AIEOP = Associazione Italiana Ematologia Oncologia Pediatrica; CALGB = Cancer and Leukemia Group B; CCG = Children's Cancer Group; DCOG = Dutch Childhood Oncology Group; FRALLE = French Acute Lymphoblastic Leukaemia; GIMEMA = Gruppo Italiano Malattie e Matologiche dell'Adulto; HOVON = Dutch-Belgian Hemato-Oncology Cooperative Group; LALA = France-Belgium Group for Lymphoblastic Acute Leukemia in Adults; MRC = Medical Research Council (United Kingdom); NOPHO = Nordic Society for Pediatric Hematology and Oncology; UKALL = United Kingdom Acute Lymphoblastic Leukaemia. | | United States[35] | | | | | CCG (Pediatric) | 197 | 16 | 67, OS 7 y | | CALGB (Adult) | 124 | 19 | 46 | | | | France[40] | | | | | FRALLE 93 (Pediatric) | 77 | 16 | 67 EFS | | LALA 94 | 100 | 18 | 41 | | | | Italy[46] | | | | | AIEOP (Pediatric) | 150 | 15 | 80, OS 2 y | | GIMEMA (Adult) | 95 | 16 | 71 | | | | Netherlands[47] | | | | | DCOG (Pediatric) | 47 | 12 | 71 EFS | | HOVON | 44 | 20 | 38 | | | | Sweden[48] | | | | | NOPHO 92 (Pediatric) | 36 | 16 | 74, OS 5 y | | Adult ALL | 99 | 18 | 39 | | | | United Kingdom[38] | | | | | MRC ALL (Pediatric) | 61 | 15-17 | 71, OS 5 y | | UKALL XII (Adult) | 67 | 15-17 | 56 | | UKALL 2003[45] | 229 | 16-24 | 72 EFS | Osteonecrosis Adolescents with ALL appear to be at higher risk than younger children for developing therapy-related complications, including osteonecrosis, deep venous thromboses, and pancreatitis.[37,49] Before the use of postinduction intensification for treatment of ALL, osteonecrosis was infrequent. The improvement in outcome for children and adolescents aged 10 years and older was accompanied by an increased incidence of osteonecrosis. The weight-bearing joints are affected in 95% of patients who develop osteonecrosis and operative interventions are needed for management of symptoms and impaired mobility in more than 40% of cases. The majority of the cases are diagnosed within the first 2 years of therapy and often the symptoms are recognized during maintenance. Evidence (osteonecrosis): - In the CCG-1961 high-risk ALL study, alternate-week dosing of dexamethasone was compared with standard continuous dexamethasone during delayed intensification to see if the osteonecrosis risk could be reduced.[49]
- The median age at symptom onset was 16 years.
- The cumulative incidence was higher in adolescents and young adults aged 16 to 21 years (20% at 5 years) than in those aged 10 to 15 years (9.9%) or in patients aged 1 to 9 years (1%).
- Operative interventions are needed for management of symptoms and impaired mobility in more than 40% of cases.
- The use of alternate-week dosing of dexamethasone as compared with standard continuous dexamethasone during delayed intensification in CCG-1961 reduced the risk of osteonecrosis. The greatest impact was seen in females aged 16 to 21 years, who showed the highest incidence of osteonecrosis with standard therapy containing continuous dexamethasone; osteonecrosis was reduced with alternate-week dexamethasone postinduction (57.6% to 5.6%).
- In the COG AALL0232 (NCT00075725) high-risk ALL trial, patients were randomly assigned during induction to receive either 14 days of dexamethasone or 28 days of prednisone.[16]
- The incidence of osteonecrosis in patients older than 10 years who received dexamethasone was 24.3%, compared with an incidence of 15.9% in those receiving prednisone (P = .001)
- Efficacy and other toxicities were comparable in the two arms.
Treatment options under clinical evaluation for adolescent and young adult patients with ALL Treatment options under clinical evaluation include the following: - NCI-2014-00712; AALL1231 (NCT02112916)(Combination Chemotherapy With or Without Bortezomib in Treating Younger Patients With Newly Diagnosed T-Cell ALL or Stage II-IV T-Cell Lymphoblastic Lymphoma): This phase III trial for patients aged 1 to 30 years with T-cell ALL is utilizing a modified augmented BFM regimen. Patients are classified into one of three risk groups (standard, intermediate, or very high) based on morphologic response at day 29, MRD status at day 29 and end of consolidation, and CNS status at diagnosis. Age and presenting leukocyte count are not used to stratify patients. The objectives of the trial include the following:
- To compare EFS in patients who are randomly assigned to receive or not to receive bortezomib on a modified augmented BFM backbone.
- To determine the safety and feasibility of modifying standard COG therapy for T-cell ALL by using dexamethasone instead of prednisone during the induction and maintenance phases and additional doses of pegaspargase during the induction and delayed intensification phases.
- To determine whether prophylactic cranial radiation can be omitted in 85% to 90% of T-cell ALL patients (non-very high risk, non-CNS3) without an increase in relapse risk, compared with historic controls.
- To determine the proportion of patients with end consolidation MRD >0.1% who become MRD-negative after intensification therapy using three high-risk BFM blocks that include high-dose cytarabine, high-dose methotrexate, ifosfamide, and etoposide.
- COG-AALL1131 (NCT01406756) (Combination Chemotherapy in Treating Young Patients With Newly Diagnosed High-Risk ALL):
This protocol is open to patients aged 30 years or younger. Patients treated on this trial are classified as high risk who lack very high-risk features and two groups of NCI standard-risk patients who otherwise lack very high-risk features: (1) those without favorable genetics (no ETV6-RUNX1 or double trisomies 4 and 10) and with day 8 peripheral blood MRD greater than 1%; and (2) those with favorable cytogenetics and high marrow MRD at day 29. Patients with BCR-ABL1 (Ph+) are treated on a separate clinical trial. Patients on this trial receive a four-drug induction (vincristine, corticosteroid, daunorubicin, and intravenous pegaspargase) with intrathecal chemotherapy. Patients younger than 10 years receive 2 weeks of dexamethasone during induction, and those aged 10 years and older receive 4 weeks of prednisone. Postinduction therapy consists of a modified augmented BFM backbone, including an interim maintenance phase with high-dose methotrexate and one delayed intensification phase. For high-risk patients, the study compares triple intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) with intrathecal methotrexate in a randomized fashion to determine whether triple intrathecal chemotherapy reduces CNS relapse rates and improves EFS. Patients aged 1 to 30 years with very high-risk features are eligible to be treated on the very high risk stratum of COG-AALL1131. The presence of any of the following features classify a patient as very high risk. - Age 13 years and older.
- CNS3 at diagnosis.
- M3 marrow at day 29.
- Unfavorable genetics (e.g., iAMP21, low hypodiploidy, MLL (KMT2A) gene rearrangements).
- High marrow MRD (>0.01% by flow cytometry) at day 29 (with the exception of NCI standard-risk patients with favorable genetics).
Very high-risk patients are treated with a BFM backbone including high-dose methotrexate during the first interim maintenance phase, a single delayed intensification phase, and a second interim maintenance phase with Capizzi methotrexate plus pegaspargase. For very high-risk patients, the study compares, in a randomized fashion, standard versus intensified therapy during the last 4 weeks of consolidation and delayed intensification. Patients are randomly assigned to receive either standard dosing of cyclophosphamide, thioguanine, and low-dose cytarabine or cyclophosphamide and etoposide. All other components of consolidation and delayed intensification are identical between arms. The trial originally included a third arm that incorporated clofarabine, cyclophosphamide, and etoposide during the last 4 weeks of consolidation and delayed intensification, but this arm was permanently closed after interim safety analyses identified that it was associated with excessive toxicity deemed to be unacceptable, including grade 4 and 5 infection and prolonged myelosuppression.
Philadelphia Chromosome-positive (BCR-ABL1-positive) ALL Philadelphia chromosome-positive (Ph+) ALL is seen in about 3% of pediatric ALL cases, increases in adolescence, and is seen in 15% to 25% of adults. In the past, this subtype of ALL has been recognized as extremely difficult to treat with poor outcome. In 2000, an international pediatric leukemia group reported a 7-year EFS of 25%, with an OS of 36%.[50] In 2010, the same group reported a 7-year EFS of 31% and an overall survival of 44% in Ph+ ALL patients treated without tyrosine kinase inhibitors.[51] Treatment of this subgroup has evolved from emphasis on aggressive chemotherapy, to bone marrow transplantation, and currently to combination therapy using chemotherapy plus a tyrosine kinase inhibitor. Treatment options Pre-tyrosine kinase inhibitor era Before the use of imatinib mesylate, HSCT from a matched sibling donor was the treatment of choice for patients with Ph+ ALL.[52] Data to support this include a retrospective multigroup analysis of children and young adults with Ph+ ALL, in which HSCT from a matched sibling donor was associated with a better outcome than standard (pre-imatinib mesylate) chemotherapy.[50] In this retrospective analysis, Ph+ ALL patients undergoing HSCT from an unrelated donor had a very poor outcome. However, in a follow-up study by the same group evaluating outcomes in the subsequent decade (pre-imatinib mesylate era), transplantation with matched-related or matched-unrelated donors were equivalent. DFS at the 5-year time point showed an advantage for transplantation in first remission compared with chemotherapy that was borderline significant (P = .049), and OS was also higher for transplantation compared with chemotherapy, although the advantage at 5 years was not significant.[51] Factors significantly associated with favorable prognosis in the pre-tyrosine kinase inhibitor era included the following: - Younger age at diagnosis.[51]
- Lower leukocyte count at diagnosis.[51]
- Early response measures.[51,53,54]
- Ph+ ALL with a rapid morphologic response or rapid peripheral blood response to induction therapy.[51,53]
Following MRD by reverse transcription polymerase chain reaction for the BCR-ABL1 fusion transcript may also be useful to help predict outcome for Ph+ patients.[55,56,57] Tyrosine kinase inhibitor era Imatinib mesylate is a selective inhibitor of the BCR-ABL protein kinase. Phase I and II studies of single-agent imatinib in children and adults with relapsed or refractory Ph+ ALL have demonstrated relatively high response rates, although these responses tended to be of short duration.[58,59] Clinical trials in adults and children with Ph+ ALL have demonstrated the feasibility of administering imatinib mesylate in combination with multiagent chemotherapy.[60,61,62] Outcome of results for Ph+ ALL demonstrated a better outcome after HSCT if imatinib was given before or after transplant.[63,64,65,66,67] Clinical trials have also demonstrated that many pediatric Ph+ ALL patients can be successfully treated without transplant using a combination of intensive chemotherapy and a tyrosine kinase inhibitor.[67,68] Evidence (tyrosine kinase inhibitor): - A retrospective study of 30 pediatric patients with Ph+ ALL (19 patients treated between 1991-2004 without a tyrosine kinase inhibitor, and 11 patients treated between 2004-2012 with either imatinib or dasatinib) indicated that tyrosine kinase inhibitors, when started mid-induction, are associated with lower end-induction MRD.[69]
- The COG-AALL0031 study evaluated whether imatinib mesylate could be incorporated into an intensive chemotherapy regimen for children with Ph+ ALL. Patients received imatinib mesylate in conjunction with chemotherapy during postinduction therapy. Some children proceeded to allogeneic HSCT after two cycles of consolidation chemotherapy with imatinib mesylate, while other patients received imatinib mesylate in combination with chemotherapy throughout all treatment phases.[62,67]
- The 5-year DFS for the 25 patients who received intensive chemotherapy with continuous dosing of imatinib mesylate was 70% ± 12%. These patients fared better than historic controls treated with chemotherapy alone (without imatinib mesylate), and at least as well as the other patients on the trial who underwent allogeneic transplantation. Five-year DFS was 66% for patients undergoing sibling-donor transplant (n = 21) and 59% for those undergoing unrelated donor transplant (n = 13).
- Patients with additional cytogenetic abnormalities had worse outcomes (P = .05).
- A nonrandomized study reported the outcome in 16 pediatric patients with Ph+ ALL who were treated with chemotherapy, imatinib, and allogeneic HSCT.[66]
- With a median follow-up of 65 months, the 5-year EFS was 81% for patients who received imatinib compared with 30% (P = .01) for a historic control group treated similarly, but without imatinib.[70] Of note, only one of the 16 patients received prophylactic imatinib posttransplant.
- The EsPhALL trial tested whether imatinib (administered discontinuously) given in the context of intensive chemotherapy improves outcome for pediatric Ph+ ALL patients, most of whom (80%) received an allogeneic HSCT in first CR. Patients were classified as either good risk or poor risk based on early response measures and remission status at the end of induction. Good-risk patients (n = 90) were randomly assigned to receive imatinib or not; poor-risk patients (n = 70) were directly assigned to imatinib. Interpretation of this study is limited due to the high noncompliance rate with randomized assignment in good-risk patients and early closure before reaching goal accrual due to publication of the results of the COG AALL0031 trial on which imatinib had been given continuously with chemotherapy.[68]
- The overall DFS of patients treated on this trial appeared to be better than historic controls, and when analyzed as-treated (and not by intent-to-treat), good-risk patients who received imatinib had a superior DFS.
- The EsPhALL trial has since been amended to test continuous dosing of imatinib; results are pending.
Dasatinib, a second-generation inhibitor of tyrosine kinases, is currently being studied in the initial treatment of Ph+ ALL. Dasatinib has shown significant activity in the CNS, both in a mouse model and a series of patients with CNS-positive leukemia.[71] The results of a phase I trial of dasatinib in pediatric patients indicated that once-daily dosing was associated with an acceptable toxicity profile, with few nonhematologic grade 3 or 4 adverse events.[72] Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with Philadelphia chromosome positive childhood precursor acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Hunger SP, Lu X, Devidas M, et al.: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 30 (14): 1663-9, 2012.
-
Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
-
Winter SS, Devidas M, Chen S, et al.: Capizzi-Style methotrexate with pegasparagase (C-MTX) is superior to high-dose methotrexate (HDMTX) in t-lineage acute lymphoblastic leukemia (T-ALL): results from Children's Oncology Group (COG) AALL0434. [Abstract] Blood 126 (23): A-794, 2015. Also available online. Last accessed January 27, 2017.
-
LeClerc JM, Billett AL, Gelber RD, et al.: Treatment of childhood acute lymphoblastic leukemia: results of Dana-Farber ALL Consortium Protocol 87-01. J Clin Oncol 20 (1): 237-46, 2002.
-
Asselin BL, Devidas M, Wang C, et al.: Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404). Blood 118 (4): 874-83, 2011.
-
Asselin BL, Devidas M, Chen L, et al.: Cardioprotection and Safety of Dexrazoxane in Patients Treated for Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia or Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma: A Report of the Children's Oncology Group Randomized Trial Pediatric Oncology Group 9404. J Clin Oncol 34 (8): 854-62, 2016.
-
Chow EJ, Asselin BL, Schwartz CL, et al.: Late Mortality After Dexrazoxane Treatment: A Report From the Children's Oncology Group. J Clin Oncol 33 (24): 2639-45, 2015.
-
Seibel NL, Asselin BL, Nachman JB, et al.: Treatment of high risk T-cell acute lymphoblastic leukemia (T-ALL): comparison of recent experience of the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG). [Abstract] Blood 104 (11): A-681, 2004.
-
Seibel NL, Steinherz PG, Sather HN, et al.: Early postinduction intensification therapy improves survival for children and adolescents with high-risk acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 111 (5): 2548-55, 2008.
-
Hastings C, Gaynon PS, Nachman JB, et al.: Increased post-induction intensification improves outcome in children and adolescents with a markedly elevated white blood cell count (≥200 × 10(9) /l) with T cell acute lymphoblastic leukaemia but not B cell disease: a report from the Children's Oncology Group. Br J Haematol 168 (4): 533-46, 2015.
-
Matloub Y, Stork L, Asselin B, et al.: Outcome of Children with Standard-Risk T-Lineage Acute Lymphoblastic Leukemia--Comparison among Different Treatment Strategies. Pediatr Blood Cancer 63 (2): 255-61, 2016.
-
Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.
-
Kurtzberg J, Ernst TJ, Keating MJ, et al.: Phase I study of 506U78 administered on a consecutive 5-day schedule in children and adults with refractory hematologic malignancies. J Clin Oncol 23 (15): 3396-403, 2005.
-
Winter SS, Dunsmore KP, Devidas M, et al.: Safe integration of nelarabine into intensive chemotherapy in newly diagnosed T-cell acute lymphoblastic leukemia: Children's Oncology Group Study AALL0434. Pediatr Blood Cancer 62 (7): 1176-83, 2015.
-
Dunsmore KP, Devidas M, Linda SB, et al.: Pilot study of nelarabine in combination with intensive chemotherapy in high-risk T-cell acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol 30 (22): 2753-9, 2012.
-
Larsen EC, Devidas M, Chen S, et al.: Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. J Clin Oncol 34 (20): 2380-8, 2016.
-
Silverman LB: Acute lymphoblastic leukemia in infancy. Pediatr Blood Cancer 49 (7 Suppl): 1070-3, 2007.
-
Hilden JM, Dinndorf PA, Meerbaum SO, et al.: Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108 (2): 441-51, 2006.
-
Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
-
Dreyer ZE, Hilden JM, Jones TL, et al.: Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer 62 (3): 419-26, 2015.
-
van der Linden MH, Valsecchi MG, De Lorenzo P, et al.: Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood 114 (18): 3764-8, 2009.
-
Tomizawa D, Koh K, Sato T, et al.: Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 21 (11): 2258-63, 2007.
-
Van der Velden VH, Corral L, Valsecchi MG, et al.: Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia 23 (6): 1073-9, 2009.
-
Salzer WL, Jones TL, Devidas M, et al.: Decreased induction morbidity and mortality following modification to induction therapy in infants with acute lymphoblastic leukemia enrolled on AALL0631: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (3): 414-8, 2015.
-
Kosaka Y, Koh K, Kinukawa N, et al.: Infant acute lymphoblastic leukemia with MLL gene rearrangements: outcome following intensive chemotherapy and hematopoietic stem cell transplantation. Blood 104 (12): 3527-34, 2004.
-
Dreyer ZE, Dinndorf PA, Camitta B, et al.: Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: a report from the Children's Oncology Group. J Clin Oncol 29 (2): 214-22, 2011.
-
Mann G, Attarbaschi A, Schrappe M, et al.: Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood 116 (15): 2644-50, 2010.
-
Kato M, Hasegawa D, Koh K, et al.: Allogeneic haematopoietic stem cell transplantation for infant acute lymphoblastic leukaemia with KMT2A (MLL) rearrangements: a retrospective study from the paediatric acute lymphoblastic leukaemia working group of the Japan Society for Haematopoietic Cell Transplantation. Br J Haematol 168 (4): 564-70, 2015.
-
Nachman J: Clinical characteristics, biologic features and outcome for young adult patients with acute lymphoblastic leukaemia. Br J Haematol 130 (2): 166-73, 2005.
-
Pui CH, Pei D, Campana D, et al.: Improved prognosis for older adolescents with acute lymphoblastic leukemia. J Clin Oncol 29 (4): 386-91, 2011.
-
Nachman JB, La MK, Hunger SP, et al.: Young adults with acute lymphoblastic leukemia have an excellent outcome with chemotherapy alone and benefit from intensive postinduction treatment: a report from the children's oncology group. J Clin Oncol 27 (31): 5189-94, 2009.
-
Pichler H, Reismüller B, Steiner M, et al.: The inferior prognosis of adolescents with acute lymphoblastic leukaemia (ALL) is caused by a higher rate of treatment-related mortality and not an increased relapse rate--a population-based analysis of 25 years of the Austrian ALL-BFM (Berlin-Frankfurt-Münster) Study Group. Br J Haematol 161 (4): 556-65, 2013.
-
Burke MJ, Gossai N, Wagner JE, et al.: Survival differences between adolescents/young adults and children with B precursor acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 19 (1): 138-42, 2013.
-
Bhatia S, Landier W, Shangguan M, et al.: Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: a report from the children's oncology group. J Clin Oncol 30 (17): 2094-101, 2012.
-
Stock W, La M, Sanford B, et al.: What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112 (5): 1646-54, 2008.
-
Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: emerging from the shadow of paediatric and adult treatment protocols. Pediatr Blood Cancer 47 (6): 748-56, 2006.
-
Barry E, DeAngelo DJ, Neuberg D, et al.: Favorable outcome for adolescents with acute lymphoblastic leukemia treated on Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol 25 (7): 813-9, 2007.
-
Ramanujachar R, Richards S, Hann I, et al.: Adolescents with acute lymphoblastic leukaemia: outcome on UK national paediatric (ALL97) and adult (UKALLXII/E2993) trials. Pediatr Blood Cancer 48 (3): 254-61, 2007.
-
Ram R, Wolach O, Vidal L, et al.: Adolescents and young adults with acute lymphoblastic leukemia have a better outcome when treated with pediatric-inspired regimens: systematic review and meta-analysis. Am J Hematol 87 (5): 472-8, 2012.
-
Boissel N, Auclerc MF, Lhéritier V, et al.: Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol 21 (5): 774-80, 2003.
-
Huguet F, Leguay T, Raffoux E, et al.: Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol 27 (6): 911-8, 2009.
-
DeAngelo DJ, Stevenson KE, Dahlberg SE, et al.: Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 29 (3): 526-34, 2015.
-
Ribera JM, Oriol A, Sanz MA, et al.: Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Español de Tratamiento en Hematología pediatric-based protocol ALL-96. J Clin Oncol 26 (11): 1843-9, 2008.
-
Hayakawa F, Sakura T, Yujiri T, et al.: Markedly improved outcomes and acceptable toxicity in adolescents and young adults with acute lymphoblastic leukemia following treatment with a pediatric protocol: a phase II study by the Japan Adult Leukemia Study Group. Blood Cancer J 4: e252, 2014.
-
Hough R, Rowntree C, Goulden N, et al.: Efficacy and toxicity of a paediatric protocol in teenagers and young adults with Philadelphia chromosome negative acute lymphoblastic leukaemia: results from UKALL 2003. Br J Haematol 172 (3): 439-51, 2016.
-
Testi AM, Valsecchi MG, Conter V, et al.: Difference in outcome of adolescents with acute lymphoblastic leukemia (ALL) enrolled in pediatric (AIEOP) and adult (GIMEMA) protocols. [Abstract] Blood 104: A-1954, 2004.
-
de Bont JM, van der Holt B, Dekker AW, et al.: [Adolescents with acute lymphatic leukaemia achieve significantly better results when treated following Dutch paediatric oncology protocols than with adult protocols]. Ned Tijdschr Geneeskd 149 (8): 400-6, 2005.
-
Hallböök H, Gustafsson G, Smedmyr B, et al.: Treatment outcome in young adults and children >10 years of age with acute lymphoblastic leukemia in Sweden: a comparison between a pediatric protocol and an adult protocol. Cancer 107 (7): 1551-61, 2006.
-
Mattano LA Jr, Devidas M, Nachman JB, et al.: Effect of alternate-week versus continuous dexamethasone scheduling on the risk of osteonecrosis in paediatric patients with acute lymphoblastic leukaemia: results from the CCG-1961 randomised cohort trial. Lancet Oncol 13 (9): 906-15, 2012.
-
Aricò M, Valsecchi MG, Camitta B, et al.: Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med 342 (14): 998-1006, 2000.
-
Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
-
Hahn T, Wall D, Camitta B, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 11 (11): 823-61, 2005.
-
Roy A, Bradburn M, Moorman AV, et al.: Early response to induction is predictive of survival in childhood Philadelphia chromosome positive acute lymphoblastic leukaemia: results of the Medical Research Council ALL 97 trial. Br J Haematol 129 (1): 35-44, 2005.
-
Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
-
Cazzaniga G, Lanciotti M, Rossi V, et al.: Prospective molecular monitoring of BCR/ABL transcript in children with Ph+ acute lymphoblastic leukaemia unravels differences in treatment response. Br J Haematol 119 (2): 445-53, 2002.
-
Jones LK, Saha V: Philadelphia positive acute lymphoblastic leukaemia of childhood. Br J Haematol 130 (4): 489-500, 2005.
-
Lee S, Kim YJ, Chung NG, et al.: The extent of minimal residual disease reduction after the first 4-week imatinib therapy determines outcome of allogeneic stem cell transplantation in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 115 (3): 561-70, 2009.
-
Champagne MA, Capdeville R, Krailo M, et al.: Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 104 (9): 2655-60, 2004.
-
Ottmann OG, Druker BJ, Sawyers CL, et al.: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 100 (6): 1965-71, 2002.
-
Thomas DA, Faderl S, Cortes J, et al.: Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 103 (12): 4396-407, 2004.
-
Yanada M, Takeuchi J, Sugiura I, et al.: High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 24 (3): 460-6, 2006.
-
Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
-
Burke MJ, Trotz B, Luo X, et al.: Allo-hematopoietic cell transplantation for Ph chromosome-positive ALL: impact of imatinib on relapse and survival. Bone Marrow Transplant 43 (2): 107-13, 2009.
-
Lee S, Kim YJ, Min CK, et al.: The effect of first-line imatinib interim therapy on the outcome of allogeneic stem cell transplantation in adults with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 105 (9): 3449-57, 2005.
-
de Labarthe A, Rousselot P, Huguet-Rigal F, et al.: Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood 109 (4): 1408-13, 2007.
-
Rives S, Estella J, Gómez P, et al.: Intermediate dose of imatinib in combination with chemotherapy followed by allogeneic stem cell transplantation improves early outcome in paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (ALL): results of the Spanish Cooperative Group SHOP studies ALL-94, ALL-99 and ALL-2005. Br J Haematol 154 (5): 600-11, 2011.
-
Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
-
Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
-
Jeha S, Coustan-Smith E, Pei D, et al.: Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 120 (10): 1514-9, 2014.
-
Rives S, Camós M, Estella J, et al.: Longer follow-up confirms major improvement in outcome in children and adolescents with Philadelphia chromosome acute lymphoblastic leukaemia treated with continuous imatinib and haematopoietic stem cell transplantation. Results from the Spanish Cooperative Study SHOP/ALL-2005. Br J Haematol 162 (3): 419-21, 2013.
-
Porkka K, Koskenvesa P, Lundán T, et al.: Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112 (4): 1005-12, 2008.
-
Zwaan CM, Rizzari C, Mechinaud F, et al.: Dasatinib in children and adolescents with relapsed or refractory leukemia: results of the CA180-018 phase I dose-escalation study of the Innovative Therapies for Children with Cancer Consortium. J Clin Oncol 31 (19): 2460-8, 2013.
Treatment of Relapsed Childhood ALLPrognostic Factors After First Relapse of Childhood ALL The prognosis for a child with acute lymphoblastic leukemia (ALL) whose disease recurs depends on multiple factors.[1,2,3,4,5,6,7,8,9,10,11,12,13,14]; [15][Level of evidence: 3iiDi] The two most important prognostic risk factors after first relapse of childhood ALL are the following: - Site of relapse.
- Time from diagnosis to relapse.
Other prognostic factors include the following: - Patient characteristics (e.g., age and peripheral blast count at time of relapse).
- Risk group classification at initial diagnosis.
- Response to reinduction therapy.
- Cytogenetics/genomic alterations.
- Immunophenotype.
Site of relapse Patients who have isolated extramedullary relapse fare better than those who have relapse involving the marrow. In some studies, patients with combined marrow/extramedullary relapse had a better prognosis than did those with a marrow relapse; however, other studies have not confirmed this finding.[5,13,16] Time from diagnosis to relapse For patients with relapsed precursor B-cell ALL, early relapses fare worse than later relapses, and marrow relapses fare worse than isolated extramedullary relapses. For example, survival rates range from less than 20% for patients with marrow relapses occurring within 18 months from diagnosis to 40% to 50% for those whose relapses occur more than 36 months from diagnosis.[5,13] For patients with isolated central nervous system (CNS) relapses, the overall survival (OS) rates for early relapse (<18 months from diagnosis) are 40% to 50% and 75% to 80% for those with late relapses (>18 months from diagnosis).[13,17] No evidence exists that early detection of relapse by frequent surveillance (complete blood counts or bone marrow tests) in off-therapy patients improves outcome.[18] Patient characteristics Age 10 years and older at diagnosis and at relapse have been reported as independent predictors of poor outcome.[13,16] A Children's Oncology Group (COG) study further showed that although patients aged 10 to 15 years at initial diagnosis do worse than patients aged 1 to 9 years (35% vs. 48%, 3-year postrelapse survival), those older than age 15 years did much worse (3-year OS, 15%; P = .001).[19] The Berlin-Frankfurt-Münster (BFM) group has also reported that high peripheral blast counts (>10,000/μL) at the time of relapse were associated with inferior outcomes in patients with late marrow relapses.[10] Children with Down syndrome with relapse of ALL have generally had inferior outcomes resulting from increased induction deaths, treatment-related mortality, and relapse. - The BFM group showed that since 2000, improvements in supportive care have led to decreases in treatment-related mortality in children with Down syndrome, but the risk of relapse remains high.[20]
- An analysis of data from the Center for International Blood and Marrow Transplant Research (CIBMTR) on 27 Down syndrome patients with ALL who underwent hematopoietic stem cell transplantation (HSCT) between 2000 and 2009 substantiated this finding. They noted that with current transplant practices, hematopoietic recovery, graft-versus-host disease (GVHD), and transplant-related mortality were within the expected range compared with non-Down syndrome ALL patients. However, relapse was higher than expected (>50%) and was the primary cause of treatment failure, leading to poor survival (24% disease-free survival [DFS] at 3 years).[21][Level of evidence: 3iiiA]
Risk group classification at initial diagnosis The COG reported that risk group classification at the time of initial diagnosis was prognostically significant after relapse; patients who met National Cancer Institute (NCI) standard-risk criteria at initial diagnosis fared better after relapse than did NCI high-risk patients.[13] Response to reinduction therapy Patients with marrow relapses who have persistent morphologic disease at the end of the first month of reinduction therapy have an extremely poor prognosis, even if they subsequently achieve a second complete remission (CR).[22][Level of evidence: 2Di]; [23][Level of evidence: 3iiiA] Several studies have demonstrated that minimal residual disease (MRD) levels after the achievement of second CR are of prognostic significance in relapsed ALL.[22,24,25,26]; [27][Level of evidence: 3iiiDi] High levels of MRD at the end of reinduction and at later time points have been correlated with an extremely high risk of subsequent relapse. Cytogenetics/genomic alterations Changes in mutation profiles from diagnosis to relapse have been identified by gene sequencing.[28,29] While oncogenic gene fusions (e.g., TCF3-PBX1, ETV6-RUNX1) are almost always observed between the time of initial diagnosis and relapse, single nucleotide variants and copy number variants may be present at diagnosis, but not at relapse, and vice versa.[28] For example, while RAS family mutations are common at both diagnosis and relapse, the specific RAS family mutations may change from diagnosis to relapse as specific leukemic subclones rise and fall during the course of treatment.[28] By contrast, relapse-specific mutations in NT5C2 (a gene involved in nucleotide metabolism) have been noted in as many as 45% of ALL cases with early relapse.[28,30,31] TP53 alterations (mutations and/or copy number alterations) are observed in approximately 11% of patients with ALL at first relapse and have been associated with an increased likelihood of persistent leukemia after initial reinduction (38.5% TP53 alteration vs. 12.5% TP53 wild-type) and poor event-free survival (EFS) (9% TP53 alteration vs. 49% TP53 wild-type).[32] Approximately one-half of the TP53 alterations were present at initial diagnosis and half were newly observed at time of relapse.[32] A second genomic alteration found to predict for poor prognosis in patients with precursor B-cell ALL in first bone marrow relapse is IKZF1 deletion.[33] The frequency of IKZF1 deletion in precursor B-cell ALL patients at first relapse patients was 33% in patients in the Acute Lymphoblastic Leukemia Relapse (ALL-REZ) BFM 2002 (NCT00114348 ) study, which was approximately twice as high as the frequency described in children at initial diagnosis of ALL.[33] RAS pathway mutations (KRAS, NRAS, FLT3, and PTPN11) are common at relapse in precursor B-cell ALL patients, and they were found in approximately 40% of patients at first relapse in one study of 206 children.[28,34] As observed at diagnosis, the frequency of RAS pathway mutations at relapse differs by cytogenetic subtype (e.g., high frequency in hyperdiploid cases and low frequency in ETV6-RUNX1 cases). The presence of RAS pathway mutations at relapse was associated with early relapse. However, presence of RAS pathway mutations at relapse was not an independent predictor of outcome. Patients with ETV6-RUNX1-positive ALL appear to have a relatively favorable prognosis at first relapse, consistent with the high percentage of such patients who relapse more than 36 months after diagnosis.[33,35] - In the ALL-REZ BFM 2002 (NCT00114348) study, an EFS of 84% ± 7% was observed for patients with ETV6-RUNX1 ALL with bone marrow relapse.[33] In this study, 94% of patients with ETV6-RUNX1 had a duration of first remission that extended at least 6 months beyond completion of their primary treatment, and on multivariate analysis, time to relapse (and not the presence of ETV6-RUNX1) was an independent predictor of outcome.
- Similarly, the 5-year OS for ETV6-RUNX1 patients enrolled on the French Acute Lymphoblastic Leukaemia (FRALLE) 93 study who relapsed at any site more than 36 months after diagnosis was 81%, and the presence of ETV6-RUNX1 was associated with a favorable survival outcome compared with other late relapsing patients.[35] However, the 3-year OS of ETV6-RUNX1 patients who experienced an early relapse (<36 months) was only 31%.
Immunophenotype Immunophenotype is an important prognostic factor at relapse. Patients with T-cell ALL who experience a marrow relapse (isolated or combined) at any time during treatment or posttreatment are less likely to achieve a second remission and long-term EFS than are patients with B-cell ALL.[5,22] Standard Treatment Options for First Bone Marrow Relapse of Childhood ALL Standard treatment options for first bone marrow relapse include the following: - Reinduction chemotherapy.
- Postreinduction therapy for patients achieving a second CR.
Reinduction chemotherapy Initial treatment of relapse consists of reinduction therapy to achieve a second CR. Using either a four-drug reinduction regimen (similar to that administered to newly diagnosed high-risk patients) or an alternative regimen including high-dose methotrexate and high-dose cytarabine, approximately 85% of patients with a marrow relapse achieve a second CR at the end of the first month of treatment.[5]; [36][Level of evidence: 2A]; [22][Level of evidence: 2Di] Patients with early marrow relapses have a lower rate of achieving a morphologic second CR (approximately 70%) than do those with late marrow relapses (approximately 95%).[22,36] Evidence (reinduction chemotherapy): - A COG study used three blocks of intensive reinduction therapy with an initial four-drug combination including doxorubicin followed by two intensive consolidation blocks before either HSCT or chemotherapy continuation.[22]
- Second remission was achieved after block 1 in 68% of patients with early relapse (<36 months from initial diagnosis) and in 96% of those with later relapse.
- Blocks 2 and 3 reduced MRD in 40 of 56 patients who were MRD-positive after block 1.
- A United Kingdom-based randomized trial of ALL patients in first relapse compared reinduction with a four-drug combination using idarubicin versus mitoxantrone.[37][Level of evidence: 1iiA]
- There was no difference in second CR rates or end-reinduction MRD levels between the two study arms.
- A significant improvement in OS in the mitoxantrone arm (69% vs. 45%, P = .007) due to decreased relapse after transplantation was reported.
The potential benefit of mitoxantrone in relapsed ALL regimens requires further investigation. - Investigators from the ALL-REZ BFM group used a six-drug reinduction approach, including high-dose methotrexate. A randomized comparison of 1 g/m2 of methotrexate versus 5 g/m2 of methotrexate with reinduction showed no advantage at the higher dose.[38]
- The combination of clofarabine, cyclophosphamide, and etoposide was reported to induce remission in 42% to 56% of patients with refractory or multiply relapsed disease.[39,40]; [41][Level of evidence: 2A]
- The combination of bortezomib plus vincristine, dexamethasone, pegaspargase, and doxorubicin was reported to induce complete response (with or without platelet recovery) in 80% of multiply relapsed patients with precursor B-cell ALL.[42][Level of evidence: 3iiiDiv] Notably, this trial did not include patients who were refractory to reinduction.
- In a study of induction therapy comprising intensive asparaginase (weekly pegaspargase or 12 doses of E.coli asparaginase) with prednisone, vincristine, and doxorubicin for patients with first relapse, the second CR rate was 86% for those receiving pegaspargase and 81% for those receiving E.coli asparaginase.[43][Level of evidence: 2Di]
Patients with relapsed T-cell ALL have much lower rates of achieving second CR with standard reinduction regimens than do patients with precursor B-cell phenotype.[22] Treatment of children with first relapse of T-cell ALL in the bone marrow with single-agent therapy using the T-cell selective agent, nelarabine, has resulted in response rates of approximately 50%.[44] The combination of nelarabine, cyclophosphamide, and etoposide has produced remissions in patients with relapsed/refractory T-cell ALL.[45] Reinduction failure is a poor prognostic factor, but subsequent attempts to obtain remission can be successful and lead to survival after HSCT. Approaches have traditionally included the use of drug combinations distinct from the first attempt at treatment; these regimens often contain newer agents under investigation in clinical trials. Although survival is progressively less likely after each attempt, two to four additional attempts are often pursued, with diminishing levels of success measured after each attempt.[46] Postreinduction therapy for patients achieving a second complete remission Early-relapsing precursor B-cell ALL For precursor B-cell patients with an early marrow relapse, allogeneic transplant from a human leukocyte antigen (HLA)-identical sibling or matched unrelated donor that is performed in second remission has been reported in most studies to result in higher leukemia-free survival than a chemotherapy approach.[7,27,47,48,49,50,51,52,53,54,55] However, even with transplantation, the survival rate for patients with early marrow relapse is less than 50%. (Refer to the Hematopoietic Stem Cell Transplantation for First and Subsequent Bone Marrow Relapse section of this summary for more information.) Late-relapsing precursor B-cell ALL For patients with a late marrow relapse of precursor B-cell ALL, a primary chemotherapy approach after achievement of second CR has resulted in survival rates of approximately 50%, and it is not clear whether allogeneic transplantation is associated with superior cure rate.[5,9,37,56,57,58]; [59][Level of evidence: 3iiA] End-reinduction MRD levels may help to identify patients with a high risk of subsequent relapse if treated with chemotherapy alone (no HSCT) in second CR. Results from one study suggest that patients with a late marrow relapse who have high end-reinduction MRD may have a better outcome if they receive an allogeneic HSCT in second CR.[60] Evidence (MRD-based risk stratification for late-relapse of precursor B-cell ALL): - In a St. Jude Children's Research Hospital study, which included 23 patients with late relapses treated with chemotherapy in second CR, the 2-year cumulative incidence of relapse was 49% for the 12 patients who were MRD-positive at the end of reinduction and 0% for the 11 patients who were MRD-negative.[24]
- In BFM studies, patients are considered to be intermediate risk if they have a late isolated marrow relapse or an early or late combined marrow/extramedullary relapse. In the ALL-REZ BFM P95/96 study from this group, end-reinduction MRD (assessed by a polymerase chain reaction-based assay) significantly predicted outcomes of children with intermediate-risk relapsed B-cell ALL treated with chemotherapy alone in second CR (no HSCT).[26]
- Patients with low MRD (<10-3) had a 10-year EFS of 73%, while those with high MRD (>10-3) had a 10-year EFS of 10%. On multivariate analysis, end-reinduction MRD was the strongest independent prognostic factor.
- In a subsequent BFM study (ALL-REZ BFM 2002 [NCT00114348]), patients with intermediate-risk relapse were allocated to allogeneic HSCT if they had high MRD at the end of the first month of treatment. Those who had low end-reinduction MRD were treated with chemotherapy only (no HSCT).[60]
- The EFS of patients with high end-reinduction MRD treated with allogeneic HSCT in second CR was 64%, which was significantly better than what had been observed on the previous P95/96 trial, during which such patients received chemotherapy without HSCT. The improvement in EFS was primarily because of a significantly lower risk of relapse in the cohort receiving HSCT in second CR (cumulative incidence of relapse, 27% on the 2002 trial compared with 59% on the P95/96 trial).
- Patients with late marrow-involved relapses and low end-reinduction MRD, treated with chemotherapy only, had a 5-year EFS of 76%, confirming the results seen in the previous P95/96 trial. However, the chemotherapy-only strategy resulted in a significantly worse outcome for patients with early-combined relapses (marrow plus extramedullary site) and low end-reinduction MRD; the 5-year EFS for these patients was only 37%. Thus, patients with early-combined relapses are no longer considered intermediate risk on BFM trials, and their treatment is not risk-stratified on the basis of end-reinduction MRD.
T-cell ALL For patients with T-cell ALL who achieved remission after bone marrow relapse, outcomes with postreinduction chemotherapy alone have generally been poor,[5] and these patients are usually treated with allogeneic HSCT in second CR, regardless of time to relapse. At 3 years, OS after allogeneic transplant for T-cell ALL in second remission was reported to be 48% and DFS was 46%.[61][Level of evidence: 3iiiA] Treatment Options for Second and Subsequent Bone Marrow Relapse Although there are no studies directly comparing chemotherapy with HSCT for patients in third or subsequent CR, because cure with chemotherapy alone is rare, transplant is generally considered a reasonable approach for those achieving remission. Long-term survival for all patients after a second relapse is particularly poor, in the range of less than 10% to 20%.[53] One of the main reasons for this is failure to obtain a third remission. In spite of numerous attempts at novel combination approaches, only about 40% of children with second relapse are able to achieve remission.[62] If these patients achieve CR, HSCT has been shown to cure 20% to 35%, with failures occurring due to high rates of relapse and transplant-related mortality.[63,64,65,66,67][Level of evidence: 3iiA] Hematopoietic Stem Cell Transplantation for First and Subsequent Bone Marrow Relapse Components of the transplantation process An expert panel review of indications for HSCT was published in 2012.[68] Components of the transplant process that have been shown to be important in improving or predicting outcome of HSCT for children with ALL include the following: - Total-body irradiation (TBI)-containing transplant preparative regimens.
- MRD detection just before transplant.
- MRD detection posttransplant.
- Donor type and HLA match.
- Role of GVHD/graft-versus-leukemia (GVL) in ALL and immune modulation after transplant to prevent relapse.
TBI-containing transplant preparative regimens For patients proceeding to allogeneic HSCT, TBI appears to be an important component of the conditioning regimen. Two retrospective studies and a randomized trial suggest that transplant conditioning regimens that include TBI produce higher cure rates than do chemotherapy-only preparative regimens.[47,69,70] Fractionated TBI (total dose, 12-14 Gy) is often combined with cyclophosphamide, etoposide, thiotepa, or a combination of these agents. Study findings with these combinations have generally resulted in similar rates of survival,[71,72,73] although one study suggested that if cyclophosphamide is used without other chemotherapy drugs, a dose of TBI in the higher range may be necessary.[74] Many standard regimens include cyclophosphamide with TBI dosing between 13.2 and 14 Gy. On the other hand, when cyclophosphamide and etoposide were used with TBI, doses above 12 Gy resulted in worse survival resulting from excessive toxicity.[72] Although some studies of non-TBI approaches have shown reasonable outcomes [75,76] and have prompted a large BFM study comparing TBI versus non-TBI regimens, TBI for all but the youngest children (age <3 or <4 years) remains the most commonly used therapy in most centers in North America.[61,66] MRD detection just before transplant Remission status at the time of transplantation has long been known to be an important predictor of outcome, with patients not in CR at HSCT having very poor survival rates.[77] Several studies have also demonstrated that the level of MRD at the time of transplant is a key risk factor in children with ALL in CR undergoing allogeneic HSCT.[25,78,79,80,81,82,83,84][Level of evidence: 3iiA]; [85][Level of evidence: 3iiB] Survival rates of patients who are MRD-positive pretransplant have been reported between 20% and 47%, compared with 60% to 88% in patients who are MRD-negative. When patients have received two to three cycles of chemotherapy in an attempt to achieve an MRD-negative remission, the benefit of further intensive therapy for achieving MRD negativity must be weighed against the potential for significant toxicity. In addition, there is not clear evidence showing that MRD positivity in a patient who has received multiple cycles of therapy is a biological disease marker for poor outcome that cannot be modified, or whether further intervention bringing such patients into an MRD negative remission will overcome this risk factor and improve survival. - In one report, 13 patients with ALL and high MRD at the time of planned transplant received an additional cycle of chemotherapy in an attempt to lower MRD before proceeding to HSCT. Ten of the 13 patients (77%) remained in CR post-HSCT, with no relapses observed in the eight patients who achieved low MRD after the additional chemotherapy cycle. In comparison, only 6 of 21 high-MRD patients (29%) who proceeded directly to HSCT without receiving additional pre-HSCT chemotherapy remained in CR.[78]
MRD detection posttransplant The presence of detectable MRD post-HSCT has been associated with an increased risk of subsequent relapse.[84,86,87,88,89] The accuracy of MRD for predicting relapse increases as time from HSCT elapses and relapse risk is also higher for patients who have higher levels of MRD detected at any given time. One study showed higher sensitivity for predicting relapse using next-generation sequencing assays than with flow cytometry, especially early after HSCT.[88] Donor type and HLA match Survival rates after matched unrelated donor and umbilical cord blood transplantation have improved significantly over the past decade and offer an outcome similar to that obtained with matched sibling donor transplants.[51,90,91,92,93]; [94,95][Level of evidence: 2A]; [96][Level of evidence: 3iiiA]; [97][Level of evidence: 3iiiDii] Rates of clinically extensive GVHD and treatment-related mortality remain higher after unrelated donor transplantation compared with matched sibling donor transplants.[52,63,90] However, there is some evidence that matched unrelated donor transplantation may yield a lower relapse rate, and National Marrow Donor Program and CIBMTR analyses have demonstrated that rates of GVHD, treatment-related mortality, and OS have improved over time.[98,99,100]; [101,102][Level of evidence: 3iiA] Another CIBMTR study suggests that outcome after one or two antigen mismatched cord blood transplants may be equivalent to that for a matched family donor or a matched unrelated donor.[103] In certain cases in which no suitable donor is found or an immediate transplant is considered crucial, a haploidentical transplant utilizing large doses of stem cells may be considered.[104] Role of GVHD/GVL in ALL and immune modulation after transplant to prevent relapse Most studies of pediatric and young adult patients that address this issue suggest an effect of both acute and chronic GVHD in decreasing relapse.[90,105,106,107] - In a COG trial of transplantation for children with ALL, grades I to III acute GVHD were associated with lower relapse risk (hazard ratio [HR], 0.4; P = .04) and better EFS (multivariate analysis, HR, 0.5; P = .02). Any effect of grade IV acute GVHD in decreasing relapse risk was obscured by a marked increase in transplant-related mortality (HR, 6.4; P = .003), while grades I to III acute GVHD had no statistically detectable effect on transplant-related mortality (HR, 0.6; P = .42).[107]
- In a multivariate model, both pretransplant MRD and acute GVHD were independent predictors of relapse, with the lowest risk of relapse observed in patients with both low pretransplant MRD and grades I to III acute GVHD.[87] For patients who did not develop acute GVHD by day 55 post-HSCT, nearly all relapses occurred between days 100 and 400 post-HSCT.
Harnessing this GVL effect, a number of approaches to prevent relapse after transplantation have been studied, including withdrawal of immune suppression or donor lymphocyte infusion and targeted immunotherapies, such as monoclonal antibodies and natural killer cell therapy.[108,109] Trials in Europe and the United States have shown that patients defined as having a high risk of relapse based upon increasing recipient chimerism (i.e., increased percentage of recipient DNA markers) can successfully undergo withdrawal of immune suppression without excessive toxicity.[110,111] - One study showed that in 46 patients with increasing recipient chimerism, the 31 patients who underwent immune suppression withdrawal, donor lymphocyte infusion, or both therapies had a 3-year EFS of 37% versus 0% in the nonintervention group (P < .001).[112]
- Other studies have shown better-than-expected rates of survival of pre-HSCT, MRD-positive patients when tapering has occurred for MRD detected after HSCT.[113]
Intrathecal medication after HSCT to prevent relapse The use of post-HSCT intrathecal chemotherapy chemoprophylaxis is controversial.[114,115,116,117] Relapse after allogeneic HSCT for relapsed ALL For patients relapsing after an allogeneic HSCT for ALL, a second ablative allogeneic HSCT may be feasible. However, many patients will be unable to undergo a second HSCT procedure because of failure to achieve remission, early toxic death, or severe organ toxicity related to salvage chemotherapy.[118] Among the highly selected group of patients able to undergo a second ablative allogeneic HSCT, approximately 10% to 30% will achieve long-term EFS.[118,119,120]; [67,121][Level of evidence: 3iiA] Prognosis is more favorable in patients with longer duration of remission after the first HSCT and in patients with CR at the time of the second HSCT.[119,120,122] In addition, one study showed an improvement in survival after second HSCT if acute GVHD occurred, especially if it had not occurred after the first transplant.[123] Reduced-intensity approaches can also cure a percentage of patients when used as a second allogeneic transplant approach, but only if patients achieve a CR confirmed by flow cytometry.[124][Level of evidence: 2A] Donor leukocyte infusion has limited benefit for patients with ALL who relapse after allogeneic HSCT.[125]; [126][Level of evidence: 3iiiA] Whether a second allogeneic transplant is necessary to treat isolated CNS and testicular relapse after HSCT is unknown. A small series has shown survival in selected patients using chemotherapy alone or chemotherapy followed by a second transplant.[127][Level of evidence: 3iA] Immunotherapeutic Approaches for Refractory ALL Immunotherapeutic approaches to the treatment of refractory ALL include monoclonal antibody therapy and chimeric antigen receptor (CAR) T-cell therapy. Monoclonal antibody therapy The following two immunotherapeutic agents have been studied for the treatment of refractory B-cell ALL: - Blinatumomab. Blinatumomab is a bispecific monoclonal antibody with one site for CD3 (T cells) and the other site for CD19 (present on most B-ALL cells). Thus, blinatumomab promotes the binding of the patient's own cytotoxic T cells to B lymphoblasts, resulting in tumor being killed. In a phase I/II trial of children younger than 18 years with relapsed/refractory B-cell ALL, 27 of 70 patients (39%) treated at the recommended phase II dose achieved a CR with single-agent blinatumomab; 52% of those achieving CR were MRD negative.[128]
- Inotuzumab. Inotuzumab is an anti-CD22 monoclonal antibody that is conjugated to calicheamicin. In a trial of adult ALL patients with relapsed/refractory B-cell ALL, CR was achieved in approximately 80% of patients.[129,130] Inotuzumab has not been extensively studied in pediatric patients with B-cell ALL and is not yet labeled for use in children.
CAR T-cell therapy CAR T-cell therapy is a new strategy under clinical investigation for the treatment of relapsed and refractory ALL in second or subsequent relapse. This strategy includes engineering T cells with a CAR that redirects T-cell specificity and function.[131] One widely utilized target of CAR-modified T cells is the CD19 antigen expressed on almost all normal B cells and most B-cell malignancies. Several clinical trials of CAR T cells targeting CD19 in relapsed/refractory ALL are being conducted, with encouraging initial results, including the following: - In pilot clinical trials conducted at the Children's Hospital of Philadelphia and the Hospital of the University of Pennsylvania, 30 children and adults (25 of whom were aged 22 years or younger) with multiply relapsed or refractory CD19-positive ALL were given T cells transduced with CD19-directed CAR lentiviral vector.[132][Level of evidence: 3iiiDi] CR was obtained in 90% of patients, including 15 of 18 patients (83%) who had previously received allogeneic SCT. The 6-month EFS rate was 67%, with most patients showing persistence of the CAR T cells and B-cell aplasia through 6 months. All 30 patients experienced some degree of cytokine-release syndrome; eight patients (27%) had severe symptomatology (need for vasopressors and/or respiratory support), which was effectively treated by the anti-interleukin-6 receptor antibody tocilizumab.
- A second report from the Pediatric Oncology Branch at the NCI described the use of a different CD-19 targeted CAR T-cell product that was prepared using a retroviral vector.[133] This CD19-CAR T-cell product induced complete responses in 70% (14 of 20) of patients (aged 1-30 years) with relapsed/refractory B-cell ALL. Persistence of CAR T cells in this study was 1 to 2 months, with recovery of normal B-cell lymphopoiesis in patients who achieved CR.
Treatment of Isolated Extramedullary Relapse With improved success in treating children with ALL, the incidence of isolated extramedullary relapse has decreased. The incidence of isolated CNS relapse is less than 5%, and testicular relapse is less than 1% to 2%.[134,135,136] As with bone marrow and mixed relapses, time from initial diagnosis to relapse is a key prognostic factor in isolated extramedullary relapses.[137] In addition, age older than 6 years at relapse was noted in one study as an adverse prognostic factor for patients with an isolated extramedullary relapse, while a second study suggested age 10 years as a better cutoff.[16,138] Of note, in the majority of children with isolated extramedullary relapses, submicroscopic marrow disease can be demonstrated using sensitive molecular techniques,[139] and successful treatment strategies must effectively control both local and systemic disease. Patients with an isolated CNS relapse who show greater than 0.01% MRD in a morphologically normal marrow have a worse prognosis (5-year EFS, 30%) than do patients with either no MRD or MRD less than 0.01% (5-year EFS, 60%).[139] Isolated CNS relapse Standard treatment options for childhood ALL that has recurred in the CNS include the following: - Systemic and intrathecal chemotherapy.
- Cranial or craniospinal radiation.
- HSCT.
While the prognosis for children with isolated CNS relapse had been quite poor in the past, aggressive systemic and intrathecal therapy followed by cranial or craniospinal radiation has improved the outlook, particularly for patients who did not receive cranial radiation during their first remission.[17,137,140,141] Evidence (chemotherapy and radiation therapy): - In a Pediatric Oncology Group (POG) study using this strategy, children who had not previously received radiation therapy and whose initial remission was 18 months or longer had a 4-year EFS rate of approximately 80%, compared with EFS rates of approximately 45% for children with CNS relapse within 18 months of diagnosis.[137]
- In a follow-up POG study, children who had not previously received radiation therapy and who had initial remission of 18 months or more were treated with intensive systemic and intrathecal chemotherapy for 1 year followed by 18 Gy of cranial radiation only.[17] The 4-year EFS was 78%. Children with an initial remission of less than 18 months also received the same chemotherapy but had craniospinal radiation (24 Gy cranial/15 Gy spinal) as in the first POG study and achieved a 4-year EFS of 52%.
A number of case series describing HSCT in the treatment of isolated CNS relapse have been published.[142,143] Although some reports have suggested a possible role for HSCT for patients with isolated CNS disease with very early relapse and T-cell disease, there is less evidence for the need for HSCT in early relapse and no evidence in late relapse. The use of transplantation to treat isolated CNS relapse occurring less than 18 months from diagnosis, especially T-cell CNS relapse, requires further study. Evidence (HSCT): - Another study compared the outcome of patients treated with either HLA-matched sibling transplants or chemoradiation therapy as in the POG studies above.[144][Level of evidence: 3iiiDii] This retrospective, registry-based study included transplantation of both early (<18 months from diagnosis) and late relapses.
- The 8-year probabilities of leukemia-free survival adjusted for age (58%) and duration of first remission (66%) were similar.
- Because of the relatively good outcome of patients with isolated CNS relapse more than 18 months from diagnosis treated with chemoradiation therapy alone (>75%), transplantation is generally not recommended by the COG for this group.
- The MRC ALLR3 trial tested intensive induction with mitoxantrone versus idarubicin in relapsed ALL patients, defining a superior outcome when mitoxantrone was used. A subanalysis of 80 patients entering the trial with isolated CNS relapse included 13 patients with very early relapse (defined as <18 months from first diagnosis), 55 patients with early relapse (defined as >18 months from initial diagnosis but within 6 months of being off therapy), and 12 patients with late relapse.[16][Level of evidence: 2A]
- Those with late relapse did very well with chemotherapy/cranial radiation therapy, with 11 of 12 patients surviving.
- Allogeneic HSCT was recommended for very early and early relapse. Sixty-six patients were alive and relapse free after the planned three induction courses. Fifty-four patients with early and very early isolated CNS relapse were eligible for protocol-recommended HSCT, and 39 (72%) patients received HSCT. Twenty-one percent of these patients relapsed, compared with a relapse rate of 71% in the group not receiving HSCT.
- Of those eligible for transplant, treatment with mitoxantrone rather than idarubicin during reinduction was associated with a survival advantage (3-year progression-free survival, 61% vs. 21%; P = .027). As in the larger trial, the major advantage from the mitoxantrone arm occurred in those receiving HSCT.[16] Low patient numbers in the very early group prohibited detailed analysis of this cohort, and rates of failure within the early group treated with chemotherapy/cranial radiation therapy are inferior to other published experiences, calling into question this chemotherapy approach to early isolated CNS relapse patients.
Isolated testicular relapse The results of treatment of isolated testicular relapse depend on the timing of the relapse. The 3-year EFS of boys with overt testicular relapse during therapy is approximately 40%; it is approximately 85% for boys with late testicular relapse.[145] Standard treatment options in North America for childhood ALL that has recurred in the testes include the following: - Chemotherapy.
- Radiation therapy.
The standard approach for treating isolated testicular relapse in North America is to administer intensive chemotherapy that includes high-dose methotrexate.[146] Patients who do not respond with a CR after induction also receive local radiation therapy. In some European clinical trial groups, orchiectomy of the involved testicle is performed instead of radiation. Biopsy of the other testicle is performed at the time of relapse to determine if additional local control (surgical removal or radiation) is to be performed. A study that looked at testicular biopsy at the end of frontline therapy failed to demonstrate a survival benefit for patients with early detection of occult disease.[147] While there are limited clinical data concerning outcome without the use of radiation therapy or orchiectomy, the use of chemotherapy (e.g., high-dose methotrexate) that may be able to achieve antileukemic levels in the testes is being tested in clinical trials. Evidence (treatment of testicular relapse [case reports]): - Dutch investigators treated five boys with a late testicular relapse with high-dose methotrexate during induction (12 g/m2) and at regular intervals during the remainder of therapy (6 g/m2) without testicular radiation. All five boys were long-term survivors.[146]
- In a small series of boys who had an isolated testicular relapse after a HSCT for a prior systemic relapse of ALL, five of seven boys had extended EFS without a second HSCT.[127][Level of evidence: 3iA]
Treatment Options Under Clinical Evaluation for Relapsed Childhood ALL Trials for ALL in first relapse Treatment options under clinical evaluation for ALL in first relapse include the following: - COG-AALL1331; NCI-2014-00631 (NCT02101853) (Risk-Stratified Randomized Phase III Testing of Blinatumomab in First Relapse of Childhood B-Lymphoblastic Leukemia [B-ALL]): This trial is evaluating whether incorporation of blinatumomab improves DFS in patients with B-cell ALL in first relapse. Blinatumomab is a bi-specific antibody that binds to the CD19 antigen, expressed on nearly all B-cell ALL cells and the CD3 antigen expressed on T cells; in this way, blinatumomab juxtaposes B-lymphoblasts with a patient's own T cells, promoting leukemia cell lysis. Patients are risk-stratified based on site of relapse (marrow-involved vs. isolated extramedullary relapse), time to relapse, and MRD status after the first month of treatment. The chemotherapy backbone for the trial is based on the United Kingdom ALLR3 regimen.[37] After the first month of treatment, high-risk and intermediate-risk patients are randomly assigned to receive either two blocks of consolidation chemotherapy or two cycles of blinatumomab. These patients will then proceed to HSCT. Low-risk patients are treated without transplant; they are randomly assigned to either a control arm based on the ALLR3 protocol or an investigational arm based on the same chemotherapy backbone and also including three cycles of blinatumomab.
- TACL 2008-002 (NCT00981799) (Trial of Nelarabine, Etoposide, and Cyclophosphamide in Relapsed T-cell ALL and T-cell Lymphoblastic Lymphoma): This trial, conducted by the Therapeutic Advances in Childhood Leukemia & Lymphoma clinical trials group, is testing the feasibility of administering nelarabine in combination with cyclophosphamide and etoposide as reinduction for patients with T-cell ALL in first relapse (as well as those who failed primary induction therapy). Doses of nelarabine and cyclophosphamide will be escalated in successive cohorts of patients to determine the maximum tolerated doses of these drugs when given in combination.
- DFCI-11-237 (NCT01523977) (Everolimus With Multiagent Reinduction Chemotherapy in Pediatric Patients With ALL): Patients in first relapse are eligible to enroll on a Dana-Farber Cancer Institute ALL Consortium trial testing the feasibility of administering everolimus, an oral mTOR inhibitor, in combination with multiagent reinduction (vincristine, prednisone, doxorubicin, intravenous pegaspargase, and intrathecal chemotherapy).
Trials for ALL in second or subsequent relapse or refractory ALL Multiple clinical trials investigating new agents and new combinations of agents are available for children with second or subsequent relapsed or refractory ALL and should be considered. These trials are testing targeted treatments specific for ALL, including monoclonal antibody-based therapies and drugs that inhibit signal transduction pathways required for leukemia cell growth and survival. Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with recurrent childhood acute lymphoblastic leukemia. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Reismüller B, Attarbaschi A, Peters C, et al.: Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria--a population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) Study Group. Br J Haematol 144 (4): 559-70, 2009.
-
Uderzo C, Conter V, Dini G, et al.: Treatment of childhood acute lymphoblastic leukemia after the first relapse: curative strategies. Haematologica 86 (1): 1-7, 2001.
-
Chessells JM, Veys P, Kempski H, et al.: Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br J Haematol 123 (3): 396-405, 2003.
-
Rivera GK, Zhou Y, Hancock ML, et al.: Bone marrow recurrence after initial intensive treatment for childhood acute lymphoblastic leukemia. Cancer 103 (2): 368-76, 2005.
-
Einsiedel HG, von Stackelberg A, Hartmann R, et al.: Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Münster Group 87. J Clin Oncol 23 (31): 7942-50, 2005.
-
Schroeder H, Garwicz S, Kristinsson J, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a population-based study of 315 patients from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Med Pediatr Oncol 25 (5): 372-8, 1995.
-
Wheeler K, Richards S, Bailey C, et al.: Comparison of bone marrow transplant and chemotherapy for relapsed childhood acute lymphoblastic leukaemia: the MRC UKALL X experience. Medical Research Council Working Party on Childhood Leukaemia. Br J Haematol 101 (1): 94-103, 1998.
-
Buchanan GR, Rivera GK, Pollock BH, et al.: Alternating drug pairs with or without periodic reinduction in children with acute lymphoblastic leukemia in second bone marrow remission: a Pediatric Oncology Group Study. Cancer 88 (5): 1166-74, 2000.
-
Rivera GK, Hudson MM, Liu Q, et al.: Effectiveness of intensified rotational combination chemotherapy for late hematologic relapse of childhood acute lymphoblastic leukemia. Blood 88 (3): 831-7, 1996.
-
Bührer C, Hartmann R, Fengler R, et al.: Peripheral blast counts at diagnosis of late isolated bone marrow relapse of childhood acute lymphoblastic leukemia predict response to salvage chemotherapy and outcome. Berlin-Frankfurt-Münster Relapse Study Group. J Clin Oncol 14 (10): 2812-7, 1996.
-
Roy A, Cargill A, Love S, et al.: Outcome after first relapse in childhood acute lymphoblastic leukaemia - lessons from the United Kingdom R2 trial. Br J Haematol 130 (1): 67-75, 2005.
-
Rizzari C, Valsecchi MG, Aricò M, et al.: Outcome of very late relapse in children with acute lymphoblastic leukemia. Haematologica 89 (4): 427-34, 2004.
-
Nguyen K, Devidas M, Cheng SC, et al.: Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia 22 (12): 2142-50, 2008.
-
Locatelli F, Schrappe M, Bernardo ME, et al.: How I treat relapsed childhood acute lymphoblastic leukemia. Blood 120 (14): 2807-16, 2012.
-
Malempati S, Gaynon PS, Sather H, et al.: Outcome after relapse among children with standard-risk acute lymphoblastic leukemia: Children's Oncology Group study CCG-1952. J Clin Oncol 25 (36): 5800-7, 2007.
-
Masurekar AN, Parker CA, Shanyinde M, et al.: Outcome of central nervous system relapses in childhood acute lymphoblastic leukaemia--prospective open cohort analyses of the ALLR3 trial. PLoS One 9 (10): e108107, 2014.
-
Barredo JC, Devidas M, Lauer SJ, et al.: Isolated CNS relapse of acute lymphoblastic leukemia treated with intensive systemic chemotherapy and delayed CNS radiation: a pediatric oncology group study. J Clin Oncol 24 (19): 3142-9, 2006.
-
Rubnitz JE, Hijiya N, Zhou Y, et al.: Lack of benefit of early detection of relapse after completion of therapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 44 (2): 138-41, 2005.
-
Freyer DR, Devidas M, La M, et al.: Postrelapse survival in childhood acute lymphoblastic leukemia is independent of initial treatment intensity: a report from the Children's Oncology Group. Blood 117 (11): 3010-5, 2011.
-
Meyr F, Escherich G, Mann G, et al.: Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with Down syndrome. Br J Haematol 162 (1): 98-106, 2013.
-
Hitzler JK, He W, Doyle J, et al.: Outcome of transplantation for acute lymphoblastic leukemia in children with Down syndrome. Pediatr Blood Cancer 61 (6): 1126-8, 2014.
-
Raetz EA, Borowitz MJ, Devidas M, et al.: Reinduction platform for children with first marrow relapse in acute lymphoblastic lymphoma. J Clin Oncol 26 (24): 3971-8, 2008.
-
von Stackelberg A, Völzke E, Kühl JS, et al.: Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer 47 (1): 90-7, 2011.
-
Coustan-Smith E, Gajjar A, Hijiya N, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia after first relapse. Leukemia 18 (3): 499-504, 2004.
-
Sramkova L, Muzikova K, Fronkova E, et al.: Detectable minimal residual disease before allogeneic hematopoietic stem cell transplantation predicts extremely poor prognosis in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 48 (1): 93-100, 2007.
-
Eckert C, von Stackelberg A, Seeger K, et al.: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 49 (6): 1346-55, 2013.
-
Paganin M, Zecca M, Fabbri G, et al.: Minimal residual disease is an important predictive factor of outcome in children with relapsed 'high-risk' acute lymphoblastic leukemia. Leukemia 22 (12): 2193-200, 2008.
-
Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
-
Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
-
Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
-
Tzoneva G, Perez-Garcia A, Carpenter Z, et al.: Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 19 (3): 368-71, 2013.
-
Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.
-
Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
-
Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
-
Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
-
Tallen G, Ratei R, Mann G, et al.: Long-term outcome in children with relapsed acute lymphoblastic leukemia after time-point and site-of-relapse stratification and intensified short-course multidrug chemotherapy: results of trial ALL-REZ BFM 90. J Clin Oncol 28 (14): 2339-47, 2010.
-
Parker C, Waters R, Leighton C, et al.: Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet 376 (9757): 2009-17, 2010.
-
von Stackelberg A, Hartmann R, Bührer C, et al.: High-dose compared with intermediate-dose methotrexate in children with a first relapse of acute lymphoblastic leukemia. Blood 111 (5): 2573-80, 2008.
-
Locatelli F, Testi AM, Bernardo ME, et al.: Clofarabine, cyclophosphamide and etoposide as single-course re-induction therapy for children with refractory/multiple relapsed acute lymphoblastic leukaemia. Br J Haematol 147 (3): 371-8, 2009.
-
Miano M, Pistorio A, Putti MC, et al.: Clofarabine, cyclophosphamide and etoposide for the treatment of relapsed or resistant acute leukemia in pediatric patients. Leuk Lymphoma 53 (9): 1693-8, 2012.
-
Hijiya N, Thomson B, Isakoff MS, et al.: Phase 2 trial of clofarabine in combination with etoposide and cyclophosphamide in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. Blood 118 (23): 6043-9, 2011.
-
Messinger YH, Gaynon PS, Sposto R, et al.: Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 120 (2): 285-90, 2012.
-
Kelly ME, Lu X, Devidas M, et al.: Treatment of relapsed precursor-B acute lymphoblastic leukemia with intensive chemotherapy: POG (Pediatric Oncology Group) study 9411 (SIMAL 9). J Pediatr Hematol Oncol 35 (7): 509-13, 2013.
-
Berg SL, Blaney SM, Devidas M, et al.: Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: a report from the Children's Oncology Group. J Clin Oncol 23 (15): 3376-82, 2005.
-
Commander LA, Seif AE, Insogna IG, et al.: Salvage therapy with nelarabine, etoposide, and cyclophosphamide in relapsed/refractory paediatric T-cell lymphoblastic leukaemia and lymphoma. Br J Haematol 150 (3): 345-51, 2010.
-
Ko RH, Ji L, Barnette P, et al.: Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol 28 (4): 648-54, 2010.
-
Eapen M, Raetz E, Zhang MJ, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with B-precursor acute lymphoblastic leukemia in a second remission: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Blood 107 (12): 4961-7, 2006.
-
Barrett AJ, Horowitz MM, Pollock BH, et al.: Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. N Engl J Med 331 (19): 1253-8, 1994.
-
Uderzo C, Valsecchi MG, Bacigalupo A, et al.: Treatment of childhood acute lymphoblastic leukemia in second remission with allogeneic bone marrow transplantation and chemotherapy: ten-year experience of the Italian Bone Marrow Transplantation Group and the Italian Pediatric Hematology Oncology Association. J Clin Oncol 13 (2): 352-8, 1995.
-
Harrison G, Richards S, Lawson S, et al.: Comparison of allogeneic transplant versus chemotherapy for relapsed childhood acute lymphoblastic leukaemia in the MRC UKALL R1 trial. MRC Childhood Leukaemia Working Party. Ann Oncol 11 (8): 999-1006, 2000.
-
Bunin N, Carston M, Wall D, et al.: Unrelated marrow transplantation for children with acute lymphoblastic leukemia in second remission. Blood 99 (9): 3151-7, 2002.
-
Borgmann A, von Stackelberg A, Hartmann R, et al.: Unrelated donor stem cell transplantation compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission: a matched-pair analysis. Blood 101 (10): 3835-9, 2003.
-
Saarinen-Pihkala UM, Heilmann C, Winiarski J, et al.: Pathways through relapses and deaths of children with acute lymphoblastic leukemia: role of allogeneic stem-cell transplantation in Nordic data. J Clin Oncol 24 (36): 5750-62, 2006.
-
Thomson B, Park JR, Felgenhauer J, et al.: Toxicity and efficacy of intensive chemotherapy for children with acute lymphoblastic leukemia (ALL) after first bone marrow or extramedullary relapse. Pediatr Blood Cancer 43 (5): 571-9, 2004.
-
Hahn T, Wall D, Camitta B, et al.: The role of cytotoxic therapy with hematopoietic stem cell transplantation in the therapy of acute lymphoblastic leukemia in children: an evidence-based review. Biol Blood Marrow Transplant 11 (11): 823-61, 2005.
-
Borgmann A, Baumgarten E, Schmid H, et al.: Allogeneic bone marrow transplantation for a subset of children with acute lymphoblastic leukemia in third remission: a conceivable alternative? Bone Marrow Transplant 20 (11): 939-44, 1997.
-
Schroeder H, Gustafsson G, Saarinen-Pihkala UM, et al.: Allogeneic bone marrow transplantation in second remission of childhood acute lymphoblastic leukemia: a population-based case control study from the Nordic countries. Bone Marrow Transplant 23 (6): 555-60, 1999.
-
van den Berg H, de Groot-Kruseman HA, Damen-Korbijn CM, et al.: Outcome after first relapse in children with acute lymphoblastic leukemia: a report based on the Dutch Childhood Oncology Group (DCOG) relapse all 98 protocol. Pediatr Blood Cancer 57 (2): 210-6, 2011.
-
Beck JC, Cao Q, Trotz B, et al.: Allogeneic hematopoietic cell transplantation outcomes for children with B-precursor acute lymphoblastic leukemia and early or late BM relapse. Bone Marrow Transplant 46 (7): 950-5, 2011.
-
Eckert C, Henze G, Seeger K, et al.: Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J Clin Oncol 31 (21): 2736-42, 2013.
-
Burke MJ, Verneris MR, Le Rademacher J, et al.: Transplant Outcomes for Children with T Cell Acute Lymphoblastic Leukemia in Second Remission: A Report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 21 (12): 2154-9, 2015.
-
Gaynon PS: Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol 131 (5): 579-87, 2005.
-
Woolfrey AE, Anasetti C, Storer B, et al.: Factors associated with outcome after unrelated marrow transplantation for treatment of acute lymphoblastic leukemia in children. Blood 99 (6): 2002-8, 2002.
-
Afify Z, Hunt L, Green A, et al.: Factors affecting the outcome of stem cell transplantation from unrelated donors for childhood acute lymphoblastic leukemia in third remission. Bone Marrow Transplant 35 (11): 1041-7, 2005.
-
Gassas A, Ishaqi MK, Afzal S, et al.: Outcome of haematopoietic stem cell transplantation for paediatric acute lymphoblastic leukaemia in third complete remission: a vital role for graft-versus-host-disease/ graft-versus-leukaemia effect in survival. Br J Haematol 140 (1): 86-9, 2008.
-
Nemecek ER, Ellis K, He W, et al.: Outcome of myeloablative conditioning and unrelated donor hematopoietic cell transplantation for childhood acute lymphoblastic leukemia in third remission. Biol Blood Marrow Transplant 17 (12): 1833-40, 2011.
-
Kato M, Horikoshi Y, Okamoto Y, et al.: Second allogeneic hematopoietic SCT for relapsed ALL in children. Bone Marrow Transplant 47 (10): 1307-11, 2012.
-
Oliansky DM, Camitta B, Gaynon P, et al.: Role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of pediatric acute lymphoblastic leukemia: update of the 2005 evidence-based review. Biol Blood Marrow Transplant 18 (4): 505-22, 2012.
-
Davies SM, Ramsay NK, Klein JP, et al.: Comparison of preparative regimens in transplants for children with acute lymphoblastic leukemia. J Clin Oncol 18 (2): 340-7, 2000.
-
Bunin N, Aplenc R, Kamani N, et al.: Randomized trial of busulfan vs total body irradiation containing conditioning regimens for children with acute lymphoblastic leukemia: a Pediatric Blood and Marrow Transplant Consortium study. Bone Marrow Transplant 32 (6): 543-8, 2003.
-
Gassas A, Sung L, Saunders EF, et al.: Comparative outcome of hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia following cyclophosphamide and total body irradiation or VP16 and total body irradiation conditioning regimens. Bone Marrow Transplant 38 (11): 739-43, 2006.
-
Tracey J, Zhang MJ, Thiel E, et al.: Transplantation conditioning regimens and outcomes after allogeneic hematopoietic cell transplantation in children and adolescents with acute lymphoblastic leukemia. Biol Blood Marrow Transplant 19 (2): 255-9, 2013.
-
Bakr M, Rasheed W, Mohamed SY, et al.: Allogeneic hematopoietic stem cell transplantation in adolescent and adult patients with high-risk T cell acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (12): 1897-904, 2012.
-
Marks DI, Forman SJ, Blume KG, et al.: A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as conditioning regimens for patients undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant 12 (4): 438-53, 2006.
-
Lee JW, Kang HJ, Kim S, et al.: Favorable outcome of hematopoietic stem cell transplantation using a targeted once-daily intravenous busulfan-fludarabine-etoposide regimen in pediatric and infant acute lymphoblastic leukemia patients. Biol Blood Marrow Transplant 21 (1): 190-5, 2015.
-
Ruggeri A, Sanz G, Bittencourt H, et al.: Comparison of outcomes after single or double cord blood transplantation in adults with acute leukemia using different types of myeloablative conditioning regimen, a retrospective study on behalf of Eurocord and the Acute Leukemia Working Party of EBMT. Leukemia 28 (4): 779-86, 2014.
-
Duval M, Klein JP, He W, et al.: Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol 28 (23): 3730-8, 2010.
-
Balduzzi A, Di Maio L, Silvestri D, et al.: Minimal residual disease before and after transplantation for childhood acute lymphoblastic leukaemia: is there any room for intervention? Br J Haematol 164 (3): 396-408, 2014.
-
Goulden N, Bader P, Van Der Velden V, et al.: Minimal residual disease prior to stem cell transplant for childhood acute lymphoblastic leukaemia. Br J Haematol 122 (1): 24-9, 2003.
-
Bader P, Kreyenberg H, Henze GH, et al.: Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: the ALL-REZ BFM Study Group. J Clin Oncol 27 (3): 377-84, 2009.
-
Leung W, Pui CH, Coustan-Smith E, et al.: Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood 120 (2): 468-72, 2012.
-
Ruggeri A, Michel G, Dalle JH, et al.: Impact of pretransplant minimal residual disease after cord blood transplantation for childhood acute lymphoblastic leukemia in remission: an Eurocord, PDWP-EBMT analysis. Leukemia 26 (12): 2455-61, 2012.
-
Bachanova V, Burke MJ, Yohe S, et al.: Unrelated cord blood transplantation in adult and pediatric acute lymphoblastic leukemia: effect of minimal residual disease on relapse and survival. Biol Blood Marrow Transplant 18 (6): 963-8, 2012.
-
Sutton R, Shaw PJ, Venn NC, et al.: Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol 168 (3): 395-404, 2015.
-
Sanchez-Garcia J, Serrano J, Serrano-Lopez J, et al.: Quantification of minimal residual disease levels by flow cytometry at time of transplant predicts outcome after myeloablative allogeneic transplantation in ALL. Bone Marrow Transplant 48 (3): 396-402, 2013.
-
Bader P, Kreyenberg H, von Stackelberg A, et al.: Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: results of the ALL-BFM-SCT 2003 trial. J Clin Oncol 33 (11): 1275-84, 2015.
-
Pulsipher MA, Langholz B, Wall DA, et al.: Risk factors and timing of relapse after allogeneic transplantation in pediatric ALL: for whom and when should interventions be tested? Bone Marrow Transplant 50 (9): 1173-9, 2015.
-
Pulsipher MA, Carlson C, Langholz B, et al.: IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 125 (22): 3501-8, 2015.
-
Liu J, Wang Y, Xu LP, et al.: Monitoring mixed lineage leukemia expression may help identify patients with mixed lineage leukemia--rearranged acute leukemia who are at high risk of relapse after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 20 (7): 929-36, 2014.
-
Locatelli F, Zecca M, Messina C, et al.: Improvement over time in outcome for children with acute lymphoblastic leukemia in second remission given hematopoietic stem cell transplantation from unrelated donors. Leukemia 16 (11): 2228-37, 2002.
-
Saarinen-Pihkala UM, Gustafsson G, Ringdén O, et al.: No disadvantage in outcome of using matched unrelated donors as compared with matched sibling donors for bone marrow transplantation in children with acute lymphoblastic leukemia in second remission. J Clin Oncol 19 (14): 3406-14, 2001.
-
Muñoz A, Diaz-Heredia C, Diaz MA, et al.: Allogeneic hemopoietic stem cell transplantation for childhood acute lymphoblastic leukemia in second complete remission-similar outcomes after matched related and unrelated donor transplant: a study of the Spanish Working Party for Blood and Marrow Transplantation in Children (Getmon). Pediatr Hematol Oncol 25 (4): 245-59, 2008.
-
Jacobsohn DA, Hewlett B, Ranalli M, et al.: Outcomes of unrelated cord blood transplants and allogeneic-related hematopoietic stem cell transplants in children with high-risk acute lymphocytic leukemia. Bone Marrow Transplant 34 (10): 901-7, 2004.
-
Kurtzberg J, Prasad VK, Carter SL, et al.: Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood 112 (10): 4318-27, 2008.
-
Peters C, Schrappe M, von Stackelberg A, et al.: Stem-cell transplantation in children with acute lymphoblastic leukemia: A prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003 trial. J Clin Oncol 33 (11): 1265-74, 2015.
-
Smith AR, Baker KS, Defor TE, et al.: Hematopoietic cell transplantation for children with acute lymphoblastic leukemia in second complete remission: similar outcomes in recipients of unrelated marrow and umbilical cord blood versus marrow from HLA matched sibling donors. Biol Blood Marrow Transplant 15 (9): 1086-93, 2009.
-
Zhang MJ, Davies SM, Camitta BM, et al.: Comparison of outcomes after HLA-matched sibling and unrelated donor transplantation for children with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant 18 (8): 1204-10, 2012.
-
Gassas A, Sung L, Saunders EF, et al.: Graft-versus-leukemia effect in hematopoietic stem cell transplantation for pediatric acute lymphoblastic leukemia: significantly lower relapse rate in unrelated transplantations. Bone Marrow Transplant 40 (10): 951-5, 2007.
-
Harvey J, Green A, Cornish J, et al.: Improved survival in matched unrelated donor transplant for childhood ALL since the introduction of high-resolution matching at HLA class I and II. Bone Marrow Transplant 47 (10): 1294-300, 2012.
-
Majhail NS, Chitphakdithai P, Logan B, et al.: Significant improvement in survival after unrelated donor hematopoietic cell transplantation in the recent era. Biol Blood Marrow Transplant 21 (1): 142-50, 2015.
-
MacMillan ML, Davies SM, Nelson GO, et al.: Twenty years of unrelated donor bone marrow transplantation for pediatric acute leukemia facilitated by the National Marrow Donor Program. Biol Blood Marrow Transplant 14 (9 Suppl): 16-22, 2008.
-
Davies SM, Wang D, Wang T, et al.: Recent decrease in acute graft-versus-host disease in children with leukemia receiving unrelated donor bone marrow transplants. Biol Blood Marrow Transplant 15 (3): 360-6, 2009.
-
Eapen M, Rubinstein P, Zhang MJ, et al.: Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369 (9577): 1947-54, 2007.
-
Klingebiel T, Handgretinger R, Lang P, et al.: Haploidentical transplantation for acute lymphoblastic leukemia in childhood. Blood Rev 18 (3): 181-92, 2004.
-
Gustafsson Jernberg A, Remberger M, Ringdén O, et al.: Graft-versus-leukaemia effect in children: chronic GVHD has a significant impact on relapse and survival. Bone Marrow Transplant 31 (3): 175-81, 2003.
-
Dini G, Zecca M, Balduzzi A, et al.: No difference in outcome between children and adolescents transplanted for acute lymphoblastic leukemia in second remission. Blood 118 (25): 6683-90, 2011.
-
Pulsipher MA, Langholz B, Wall DA, et al.: The addition of sirolimus to tacrolimus/methotrexate GVHD prophylaxis in children with ALL: a phase 3 Children's Oncology Group/Pediatric Blood and Marrow Transplant Consortium trial. Blood 123 (13): 2017-25, 2014.
-
Pulsipher MA, Bader P, Klingebiel T, et al.: Allogeneic transplantation for pediatric acute lymphoblastic leukemia: the emerging role of peritransplantation minimal residual disease/chimerism monitoring and novel chemotherapeutic, molecular, and immune approaches aimed at preventing relapse. Biol Blood Marrow Transplant 15 (1 Suppl): 62-71, 2008.
-
Lankester AC, Bierings MB, van Wering ER, et al.: Preemptive alloimmune intervention in high-risk pediatric acute lymphoblastic leukemia patients guided by minimal residual disease level before stem cell transplantation. Leukemia 24 (8): 1462-9, 2010.
-
Horn B, Soni S, Khan S, et al.: Feasibility study of preemptive withdrawal of immunosuppression based on chimerism testing in children undergoing myeloablative allogeneic transplantation for hematologic malignancies. Bone Marrow Transplant 43 (6): 469-76, 2009.
-
Pochon C, Oger E, Michel G, et al.: Follow-up of post-transplant minimal residual disease and chimerism in childhood lymphoblastic leukaemia: 90 d to react. Br J Haematol 169 (2): 249-61, 2015.
-
Bader P, Kreyenberg H, Hoelle W, et al.: Increasing mixed chimerism is an important prognostic factor for unfavorable outcome in children with acute lymphoblastic leukemia after allogeneic stem-cell transplantation: possible role for pre-emptive immunotherapy? J Clin Oncol 22 (9): 1696-705, 2004.
-
Gandemer V, Pochon C, Oger E, et al.: Clinical value of pre-transplant minimal residual disease in childhood lymphoblastic leukaemia: the results of the French minimal residual disease-guided protocol. Br J Haematol 165 (3): 392-401, 2014.
-
Rubin J, Vettenranta K, Vettenranta J, et al.: Use of intrathecal chemoprophylaxis in children after SCT and the risk of central nervous system relapse. Bone Marrow Transplant 46 (3): 372-8, 2011.
-
Thompson CB, Sanders JE, Flournoy N, et al.: The risks of central nervous system relapse and leukoencephalopathy in patients receiving marrow transplants for acute leukemia. Blood 67 (1): 195-9, 1986.
-
Oshima K, Kanda Y, Yamashita T, et al.: Central nervous system relapse of leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 14 (10): 1100-7, 2008.
-
Ruutu T, Corradini P, Gratwohl A, et al.: Use of intrathecal prophylaxis in allogeneic haematopoietic stem cell transplantation for malignant blood diseases: a survey of the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant 35 (2): 121-4, 2005.
-
Mehta J, Powles R, Treleaven J, et al.: Outcome of acute leukemia relapsing after bone marrow transplantation: utility of second transplants and adoptive immunotherapy. Bone Marrow Transplant 19 (7): 709-19, 1997.
-
Eapen M, Giralt SA, Horowitz MM, et al.: Second transplant for acute and chronic leukemia relapsing after first HLA-identical sibling transplant. Bone Marrow Transplant 34 (8): 721-7, 2004.
-
Bosi A, Laszlo D, Labopin M, et al.: Second allogeneic bone marrow transplantation in acute leukemia: results of a survey by the European Cooperative Group for Blood and Marrow Transplantation. J Clin Oncol 19 (16): 3675-84, 2001.
-
Nishikawa T, Inagaki J, Nagatoshi Y, et al.: The second therapeutic trial for children with hematological malignancies who relapsed after their first allogeneic SCT: long-term outcomes. Pediatr Transplant 16 (7): 722-8, 2012.
-
Bajwa R, Schechter T, Soni S, et al.: Outcome of children who experience disease relapse following allogeneic hematopoietic SCT for hematologic malignancies. Bone Marrow Transplant 48 (5): 661-5, 2013.
-
Schechter T, Avila L, Frangoul H, et al.: Effect of acute graft-versus-host disease on the outcome of second allogeneic hematopoietic stem cell transplant in children. Leuk Lymphoma 54 (1): 105-9, 2013.
-
Pulsipher MA, Boucher KM, Wall D, et al.: Reduced-intensity allogeneic transplantation in pediatric patients ineligible for myeloablative therapy: results of the Pediatric Blood and Marrow Transplant Consortium Study ONC0313. Blood 114 (7): 1429-36, 2009.
-
Collins RH Jr, Goldstein S, Giralt S, et al.: Donor leukocyte infusions in acute lymphocytic leukemia. Bone Marrow Transplant 26 (5): 511-6, 2000.
-
Levine JE, Barrett AJ, Zhang MJ, et al.: Donor leukocyte infusions to treat hematologic malignancy relapse following allo-SCT in a pediatric population. Bone Marrow Transplant 42 (3): 201-5, 2008.
-
Bhadri VA, McGregor MR, Venn NC, et al.: Isolated testicular relapse after allo-SCT in boys with ALL: outcome without second transplant. Bone Marrow Transplant 45 (2): 397-9, 2010.
-
von Stackelberg A, Locatelli F, Zugmaier G, et al.: Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol 34 (36): 4381-4389, 2016.
-
Kantarjian H, Thomas D, Jorgensen J, et al.: Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 119 (15): 2728-36, 2013.
-
Kantarjian HM, DeAngelo DJ, Stelljes M, et al.: Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 375 (8): 740-53, 2016.
-
Grupp SA, Kalos M, Barrett D, et al.: Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368 (16): 1509-18, 2013.
-
Maude SL, Frey N, Shaw PA, et al.: Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371 (16): 1507-17, 2014.
-
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al.: T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385 (9967): 517-28, 2015.
-
Möricke A, Zimmermann M, Reiter A, et al.: Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM study group from 1981 to 2000. Leukemia 24 (2): 265-84, 2010.
-
Silverman LB, Stevenson KE, O'Brien JE, et al.: Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985-2000). Leukemia 24 (2): 320-34, 2010.
-
Pui CH, Pei D, Sandlund JT, et al.: Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 371-82, 2010.
-
Ritchey AK, Pollock BH, Lauer SJ, et al.: Improved survival of children with isolated CNS relapse of acute lymphoblastic leukemia: a pediatric oncology group study . J Clin Oncol 17 (12): 3745-52, 1999.
-
Domenech C, Mercier M, Plouvier E, et al.: First isolated extramedullary relapse in children with B-cell precursor acute lymphoblastic leukaemia: results of the Cooprall-97 study. Eur J Cancer 44 (16): 2461-9, 2008.
-
Hagedorn N, Acquaviva C, Fronkova E, et al.: Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of "isolated" and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM study group. Blood 110 (12): 4022-9, 2007.
-
Ribeiro RC, Rivera GK, Hudson M, et al.: An intensive re-treatment protocol for children with an isolated CNS relapse of acute lymphoblastic leukemia. J Clin Oncol 13 (2): 333-8, 1995.
-
Kumar P, Kun LE, Hustu HO, et al.: Survival outcome following isolated central nervous system relapse treated with additional chemotherapy and craniospinal irradiation in childhood acute lymphoblastic leukemia. Int J Radiat Oncol Biol Phys 31 (3): 477-83, 1995.
-
Yoshihara T, Morimoto A, Kuroda H, et al.: Allogeneic stem cell transplantation in children with acute lymphoblastic leukemia after isolated central nervous system relapse: our experiences and review of the literature. Bone Marrow Transplant 37 (1): 25-31, 2006.
-
Harker-Murray PD, Thomas AJ, Wagner JE, et al.: Allogeneic hematopoietic cell transplantation in children with relapsed acute lymphoblastic leukemia isolated to the central nervous system. Biol Blood Marrow Transplant 14 (6): 685-92, 2008.
-
Eapen M, Zhang MJ, Devidas M, et al.: Outcomes after HLA-matched sibling transplantation or chemotherapy in children with acute lymphoblastic leukemia in a second remission after an isolated central nervous system relapse: a collaborative study of the Children's Oncology Group and the Center for International Blood and Marrow Transplant Research. Leukemia 22 (2): 281-6, 2008.
-
Wofford MM, Smith SD, Shuster JJ, et al.: Treatment of occult or late overt testicular relapse in children with acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 10 (4): 624-30, 1992.
-
van den Berg H, Langeveld NE, Veenhof CH, et al.: Treatment of isolated testicular recurrence of acute lymphoblastic leukemia without radiotherapy. Report from the Dutch Late Effects Study Group. Cancer 79 (11): 2257-62, 1997.
-
Trigg ME, Steinherz PG, Chappell R, et al.: Early testicular biopsy in males with acute lymphoblastic leukemia: lack of impact on subsequent event-free survival. J Pediatr Hematol Oncol 22 (1): 27-33, 2000 Jan-Feb.
Changes to this Summary (04 / 14 / 2017)The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above. Risk-Based Treatment Assignment Added text to state that some investigators have suggested that the definition of induction failure should be expanded to include end-of-induction minimal residual disease (MRD) of more than 5%, regardless of morphologic findings (cited O'Connor et al. as reference 256). Added text about the 16-001 (NCT03020030) trial as a prognostic risk group under clinical evaluation. Treatment for Newly Diagnosed Childhood ALL Added text to state that measurement of serum asparaginase activity (SAA) levels after a mild or questionable reaction to pegaspargase may help to differentiate patients for whom the switch to Erwinia is indicated (because of inadequate SAA) from patients for whom a change in preparation may not be necessary (cited van der Sluis et al. and Bleyer et al. as references 16 and 17, respectively). Added text about several studies that have identified a subset of patients who experience silent inactivation of asparaginase, defined as absence of therapeutic SAA levels without overt allergy (cited Tong et al. and Vrooman et al. as references 18 and 19, respectively). Postinduction Treatment for Childhood ALL Added text to state that the authors also found that self-reporting was not a reliable measure of adherence, with 84% of patients over reporting compliance with taking mercaptopurine at least some of the time. The data suggest that additional measures of adherence besides self-reporting are needed (cited Landier et al. as reference 69). Revised text about the COG-AALL0932 trial for standard-risk ALL. Revised text about the COG-AALL1131 trial for high-risk and very high-risk ALL. Added text about the COG-AALL1521 trial as a treatment option under clinical evaluation for high-risk and very high-risk ALL. Added text about the 16-001 (NCT03020030) trial as another treatment option under clinical evaluation. This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages. About This PDQ SummaryPurpose of This Summary This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood acute lymphoblastic leukemia. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions. Reviewers and Updates This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH). Board members review recently published articles each month to determine whether an article should: - be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary. The lead reviewers for Childhood Acute Lymphoblastic Leukemia Treatment are: - Alan Scott Gamis, MD, MPH (Children's Mercy Hospital)
- Karen J. Marcus, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Michael A. Pulsipher, MD (Children's Hospital Los Angeles)
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Lewis B. Silverman, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries. Levels of Evidence Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations. Permission to Use This Summary PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]." The preferred citation for this PDQ summary is: PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Acute Lymphoblastic Leukemia Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/leukemia/hp/child-all-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389206] Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images. Disclaimer Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page. Contact Us More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us. Last Revised: 2017-04-14 Last modified on: 8 September 2017
|
|