Melanoma Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Melanoma Treatment (PDQ®): Treatment - Health Professional Information [NCI]Skip to the navigationGeneral Information About MelanomaMelanoma is a malignant tumor of melanocytes, which are the cells that make the pigment melanin and are derived from the neural crest. Although most melanomas arise in the skin, they may also arise from mucosal surfaces or at other sites to which neural crest cells migrate, including the uveal tract. Uveal melanomas differ significantly from cutaneous melanoma in incidence, prognostic factors, molecular characteristics, and treatment. (Refer to the PDQ summary on Intraocular (Uveal) Melanoma Treatment for more information.) Incidence and Mortality Estimated new cases and deaths from melanoma in the United States in 2017:[1] - New cases: 87,110.
- Deaths: 9,730.
Skin cancer is the most common malignancy diagnosed in the United States, with 3.5 million cancers diagnosed in 2 million people annually.[2] Melanoma represents less than 5% of skin cancers but results in most deaths.[2,3] The incidence has been increasing over the past four decades.[2] Elderly men are at highest risk; however, melanoma is the most common cancer in young adults aged 25 to 29 years and the second most common cancer in those aged 15 to 29 years.[4] Ocular melanoma is the most common cancer of the eye, with approximately 2,000 cases diagnosed annually. Risk Factors Risk factors for melanoma include both intrinsic (genetic and phenotype) and extrinsic (environmental or exposure) factors: - Sun exposure.
- Pigmentary characteristics.
- Multiple nevi.
- Family and personal history of melanoma.
- Immunosuppression.
- Environmental exposures.
(Refer to the PDQ summaries on Skin Cancer Prevention and the Genetics of Skin Cancer for more information about risk factors.) Anatomy
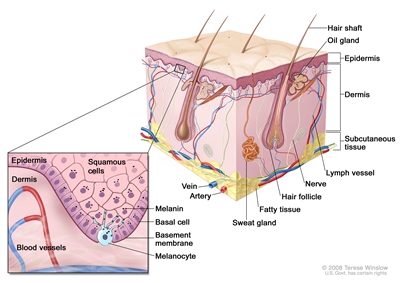
Schematic representation of normal skin. Melanocytes are also present in normal skin and serve as the source cell for melanoma. The relatively avascular epidermis houses both basal cell keratinocytes and squamous epithelial keratinocytes, the source cells for basal cell carcinoma and squamous cell carcinoma, respectively. The separation between epidermis and dermis occurs at the basement membrane zone, located just inferior to the basal cell keratinocytes.
Screening Refer to the PDQ summary on Skin Cancer Screening for more information. Clinical Features Melanoma occurs predominantly in adults, and more than 50% of the cases arise in apparently normal areas of the skin. Although melanoma can occur anywhere, including on mucosal surfaces and the uvea, melanoma in women occurs more commonly on the extremities, and in men it occurs most commonly on the trunk or head and neck.[5] Early signs in a nevus that would suggest a malignant change include the following: - Darker or variable discoloration.
- Itching.
- An increase in size or the development of satellites.
- Ulceration or bleeding (later signs).
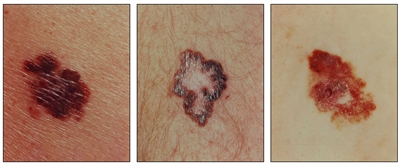
Melanomas with characteristic asymmetry, border irregularity, color variation, and large diameter.
Diagnosis A biopsy, preferably by local excision, should be performed for any suspicious lesions. Suspicious lesions should never be shaved off or cauterized. The specimens should be examined by an experienced pathologist to allow for microstaging. Studies show that distinguishing between benign pigmented lesions and early melanomas can be difficult, and even experienced dermatopathologists can have differing opinions. To reduce the possibility of misdiagnosis for an individual patient, a second review by an independent qualified pathologist should be considered.[6,7] Agreement between pathologists in the histologic diagnosis of melanomas and benign pigmented lesions has been studied and found to be considerably variable.[6,7] Evidence (discordance in histologic evaluation): - One study found that there was discordance on the diagnosis of melanoma versus benign lesions in 37 of 140 cases examined by a panel of experienced dermatopathologists. For the histologic classification of cutaneous melanoma, the highest concordance was attained for Breslow thickness and presence of ulceration, while the agreement was poor for other histologic features such as Clark level of invasion, presence of regression, and lymphocytic infiltration.[6]
- In another study, 38% of cases examined by a panel of expert pathologists had two or more discordant interpretations.[7]
Prognostic Factors Prognosis is affected by the characteristics of primary and metastatic tumors. The most important prognostic factors have been incorporated into the revised 2009 American Joint Committee on Cancer staging and include the following:[5,8,9,10] - Thickness and/or level of invasion of the melanoma.
- Mitotic index, defined as mitoses per millimeter.
- Ulceration or bleeding at the primary site.
- Number of regional lymph nodes involved, with distinction of macrometastasis and micrometastasis.
- Systemic metastasis.
- Site-nonvisceral versus lung versus all other visceral sites.
- Elevated serum lactate dehydrogenase level.
Patients who are younger, who are female, and who have melanomas on their extremities generally have better prognoses.[5,8,9,10] Microscopic satellites, recorded as present or absent, in stage I melanoma may be a poor prognostic histologic factor, but this is controversial.[11] The presence of tumor infiltrating lymphocytes, which may be categorized as brisk, nonbrisk, or absent, is under study as a potential prognostic factor.[12] The risk of relapse decreases substantially over time, although late relapses are not uncommon.[13,14] Related Summaries Other PDQ summaries containing information related to melanoma include the following: - Skin Cancer Prevention
- Skin Cancer Screening
- Skin Cancer Treatment
- Intraocular (Uveal) Melanoma Treatment
References:
-
American Cancer Society: Cancer Facts and Figures 2017. Atlanta, Ga: American Cancer Society, 2017. Available online. Last accessed May 25, 2017.
-
American Cancer Society: Cancer Facts and Figures 2014. Atlanta, Ga: American Cancer Society, 2014. Available online. Last accessed July 11, 2016.
-
Melanoma. Bethesda, Md: National Library of Medicine, 2012. Available online. Last accessed December 8, 2016.
-
Bleyer A, O'Leary M, Barr R, et al., eds.: Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975-2000. Bethesda, Md: National Cancer Institute, 2006. NIH Pub. No. 06-5767. Also available online. Last accessed January 27, 2017.
-
Slingluff CI Jr, Flaherty K, Rosenberg SA, et al.: Cutaneous melanoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1643-91.
-
Corona R, Mele A, Amini M, et al.: Interobserver variability on the histopathologic diagnosis of cutaneous melanoma and other pigmented skin lesions. J Clin Oncol 14 (4): 1218-23, 1996.
-
Farmer ER, Gonin R, Hanna MP: Discordance in the histopathologic diagnosis of melanoma and melanocytic nevi between expert pathologists. Hum Pathol 27 (6): 528-31, 1996.
-
Balch CM, Soong S, Ross MI, et al.: Long-term results of a multi-institutional randomized trial comparing prognostic factors and surgical results for intermediate thickness melanomas (1.0 to 4.0 mm). Intergroup Melanoma Surgical Trial. Ann Surg Oncol 7 (2): 87-97, 2000.
-
Manola J, Atkins M, Ibrahim J, et al.: Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol 18 (22): 3782-93, 2000.
-
Balch CM, Gershenwald JE, Soong SJ, et al.: Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27 (36): 6199-206, 2009.
-
León P, Daly JM, Synnestvedt M, et al.: The prognostic implications of microscopic satellites in patients with clinical stage I melanoma. Arch Surg 126 (12): 1461-8, 1991.
-
Mihm MC Jr, Clemente CG, Cascinelli N: Tumor infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest 74 (1): 43-7, 1996.
-
Shen P, Guenther JM, Wanek LA, et al.: Can elective lymph node dissection decrease the frequency and mortality rate of late melanoma recurrences? Ann Surg Oncol 7 (2): 114-9, 2000.
-
Tsao H, Cosimi AB, Sober AJ: Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer 79 (12): 2361-70, 1997.
Cellular and Molecular Classification of MelanomaThe descriptive terms for clinicopathologic cellular subtypes of malignant melanoma should be considered of historic interest only; they do not have independent prognostic or therapeutic significance. The cellular subtypes are the following: - Superficial spreading.
- Nodular.
- Lentigo maligna.
- Acral lentiginous (palmar/plantar and subungual).
- Miscellaneous unusual types:
- Mucosal lentiginous (oral and genital).
- Desmoplastic.
- Verrucous.
Identification of activating mutations in the mitogen-activated protein kinase pathway served as a catalyst to develop a molecular classification of melanoma. Such a classification provides potential drug targets, directions for future clinical trials, and the ability to select patients for targeted therapies. BRAF gene mutations BRAF (V-raf murine sarcoma viral oncogene homolog B1) genes, first reported in 2002, are the most frequent mutations in cutaneous melanoma. Approximately 40% to 60% of malignant melanomas harbor a single nucleotide transversion in BRAF. Most have a mutation that results in a substitution from valine to glutamic acid at position 600 (BRAF V600E); less frequent mutations include valine 600 to lysine or arginine residues (V600K/R).[1] Drugs that target this mutation by inhibiting BRAF are under evaluation in clinical trials. Vemurafenib was approved by the U.S. Food and Drug Administration (FDA) in 2011 for the treatment of unresectable or metastatic melanoma in patients who test positive for the BRAF mutation, as detected by an FDA-approved test (e.g., cobas 4800 BRAF V600 Mutation Test). Other gene mutations In smaller subsets of cutaneous melanoma, other activating mutations have been described, including the following: - NRAS (neuroblastoma RAS viral [v-ras] oncogene homolog): Approximately 15% to 20% of melanomas harbor an oncogenic NRAS mutation.[2,3]
- c-KIT: A c-KIT mutation, or increased copy number, is associated with mucosal and acral melanomas (which comprise 6%-7% of melanomas in whites but are the most common subtype in the Asian population).[4,5,6]
- CDK4 (cyclin-dependent kinase 4): CDK4 mutations have been described in approximately 4% of melanomas and are also more common in acral and mucosal melanomas.[7,8]
Drugs developed to target these mutations are currently in clinical trials. Additional oncogenes and tumor-suppressor gene candidates currently under evaluation include P13K, AKT, P53, PTEN, mTOR, Bcl-2, and MITF. Uveal melanoma Uveal melanomas differ significantly from cutaneous melanomas; in one series, 83% of 186 uveal melanomas were found to have a constitutively active somatic mutation in GNAQ or GNA11.[9,10] (Refer to the PDQ summary on Intraocular (Uveal) Melanoma Treatment for more information.) References:
-
Pollock PM, Meltzer PS: A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell 2 (1): 5-7, 2002.
-
Edlundh-Rose E, Egyházi S, Omholt K, et al.: NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res 16 (6): 471-8, 2006.
-
Goel VK, Lazar AJ, Warneke CL, et al.: Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol 126 (1): 154-60, 2006.
-
Hodi FS, Friedlander P, Corless CL, et al.: Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046-51, 2008.
-
Guo J, Si L, Kong Y, et al.: Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29 (21): 2904-9, 2011.
-
Carvajal RD, Antonescu CR, Wolchok JD, et al.: KIT as a therapeutic target in metastatic melanoma. JAMA 305 (22): 2327-34, 2011.
-
Curtin JA, Fridlyand J, Kageshita T, et al.: Distinct sets of genetic alterations in melanoma. N Engl J Med 353 (20): 2135-47, 2005.
-
Stark M, Hayward N: Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res 67 (6): 2632-42, 2007.
-
Van Raamsdonk CD, Bezrookove V, Green G, et al.: Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457 (7229): 599-602, 2009.
-
Van Raamsdonk CD, Griewank KG, Crosby MB, et al.: Mutations in GNA11 in uveal melanoma. N Engl J Med 363 (23): 2191-9, 2010.
Stage Information for MelanomaClinical staging is based on whether the tumor has spread to regional lymph nodes or distant sites. For melanoma that is clinically confined to the primary site, the chance of lymph node or systemic metastases increases as the thickness and depth of local invasion increases, which worsens the prognosis. Melanoma can spread by local extension (through lymphatics) and/or by hematogenous routes to distant sites. Any organ may be involved by metastases, but lungs and liver are common sites. The microstage of malignant melanoma is determined on histologic examination by the vertical thickness of the lesion in millimeters (Breslow classification) and/or the anatomic level of local invasion (Clark classification). The Breslow thickness is more reproducible and more accurately predicts subsequent behavior of malignant melanoma in lesions thicker than 1.5 mm and should always be reported. Accurate microstaging of the primary tumor requires careful histologic evaluation of the entire specimen by an experienced pathologist. Clark Classification (Level of Invasion) Table 1. Clark Classification (Level of Invasion)| Level of Invasion | Description |
|---|
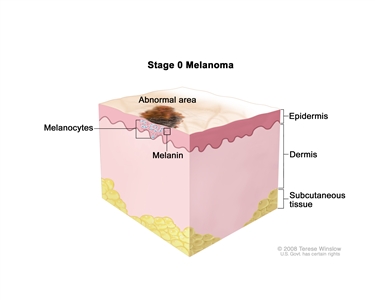
| Level I | Lesions involving only the epidermis (in situ melanoma); not an invasive lesion. | | Level II | Invasion of the papillary dermis; does not reach the papillary-reticular dermal interface. | | Level III | Invasion fills and expands the papillary dermis but does not penetrate the reticular dermis. | | Level IV | Invasion into the reticular dermis but not into the subcutaneous tissue. | | Level V | Invasion through the reticular dermis into the subcutaneous tissue. | American Joint Committee on Cancer (AJCC) Stage Groupings and TNM Definitions Melanoma staging is defined by the AJCC's TNM classification system.[1] Table 2. TNM Definitions for Stage 0 Melanoma| Stage | TNM | Description | Image |
|---|
| Clinicala | Pathologicalb | | |
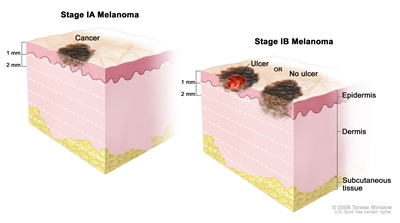
| | 0 | 0 | Tis | Melanomain situ | | N0 | No regional metastases | | M0 | No detectable evidence of distant metastases | | M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | The explanations for superscripts a and b are at the end of Table 7. | Table 3. TNM Definitions for Stage I Melanoma| Stage | TNM | Description | Image |
|---|
| M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | The explanations for superscripts a and b are at the end of Table 7. | | Clinicala | Pathologicalb | | |
| | IA | IA | T1a | Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | N0 | No regional metastases detected | | M0 | No detectable evidence of distant metastases | | IB | IB | T1b | Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a | Melanomas 1.01-2.0 mm in thickness without ulceration | | N0 | No regional metastases detected | | M0 | No detectable evidence of distant metastases | Table 4. TNM Definitions for Stage II Melanoma| Stage | TNM | Description | Image |
|---|
| Clinicala | Pathologicalb | | |
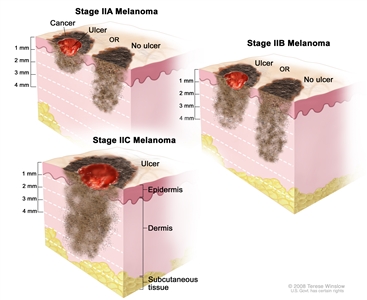
| | IIA | IIA | T2b | Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a | Melanomas 2.01-4.0 mm in thickness without ulceration | | N0 | No regional metastases detected | | M0 | No detectable evidence of distant metastases | | IIB | IIB | T3b | Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a | Melanomas >4.0 mm in thickness without ulceration | | N0 | No regional metastases detected | | M0 | No detectable evidence of distant metastases | | IIC | IIC | T4b | Melanomas >4.0 mm in thickness with ulceration | | N0 | No regional metastases detected | | M0 | No detectable evidence of distant metastases | | M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | The explanations for superscripts a and b are at the end of Table 7. | Table 5. TNM Definitions for Stage III Melanoma: Clinical Staginga| Stage | TNM | Description | Image |
|---|
| III | Any T | TX = Primary tumor cannot be assessed (e.g., curettaged or severely regressed melanoma) |
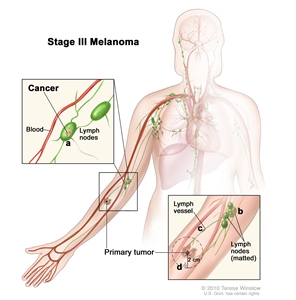
| | T0 = No evidence of primary tumor | | Tis = Melanomain situ | | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | T4b = Melanomas >4.0 mm in thickness with ulceration | | ≥N1 | N1 = 1 regional lymph node metastasis | | N2 = 2-3 regional lymph node metastases | | N3 = ≥4 regional lymph node metastases; or matted nodes; or in transit met(s)/satellite(s) with metastatic lymph node(s) | | M0 | No detectable evidence of distant metastases | | M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | The explanation for superscript a is at the end of Table 7. | Table 6. TNM Definitions for Stage III Melanoma: Pathologic Stagingb| Stage | TNM | Description |
|---|
| IIIA | T1-4a | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | N1a | 1 regional lymph node metastasis with micrometastasisc | | N2a | 2-3 regional lymph node metastases with micrometastasisc | | M0 | No detectable evidence of distant metastases | | IIIB | T1-4b | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | T4b = Melanomas >4.0 mm in thickness with ulceration | | N1a | 1 regional lymph node metastasis with micrometastasisc | | N2a | 2-3 regional lymph node metastases with micrometastasisc | | M0 | No detectable evidence of distant metastases | | IIIB | T1-4a | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | N1b | N1b = 1 regional lymph node metastasis with macrometastasisd | | N2b | N2b = 2-3 regional lymph node metastases with macrometastasisd | | N2c | N2c = In transit met(s)/satellite(s) without metastatic lymph nodes | | M0 | No detectable evidence of distant metastases | | IIIC | T1-4b | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | T4b = Melanomas >4.0 mm in thickness with ulceration | | N1b | N1b = 1 regional lymph node metastasis with macrometastasisd | | N2b | N2b = 2-3 regional lymph node metastases with macrometastasisd | | N2c | N2c = In transit met(s)/satellite(s) without metastatic lymph nodes | | M0 | No detectable evidence of distant metastases | | IIIC | Any T | TX = Primary tumor cannot be assessed (e.g., curettaged or severely regressed melanoma) | | T0 = No evidence of primary tumor | | Tis = Melanomain situ | | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration. | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | T4b = Melanomas >4.0 mm in thickness with ulceration | | N3 | ≥4 regional lymph node metastases; or matted nodes; or in transit met(s)/satellite(s) with metastatic lymph node(s) | | M0 | No detectable evidence of distant metastases | | M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | The explanations for superscripts b-d are at the end of Table 7. | Table 7. TNM Definitions for Stage IV Melanoma| Stage | TNM | Description | Image |
|---|
| Clinicala | Pathologicalb | | |
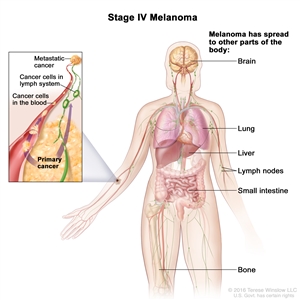
| | IV | IV | Any T | TX = Primary tumor cannot be assessed (e.g., curettaged or severely regressed melanoma) | | T0 = No evidence of primary tumor | | Tis = Melanomain situ | | T1a = Melanomas ≤1.0 mm in thickness without ulceration; mitosis <1/mm2 | | T1b = Melanomas ≤1.0 mm in thickness with ulceration or mitoses ≥1/mm2 | | T2a = Melanomas 1.01-2.0 mm in thickness without ulceration | | T2b = Melanomas 1.01-2.0 mm in thickness with ulceration | | T3a = Melanomas 2.01-4.0 mm in thickness without ulceration | | T3b = Melanomas 2.01-4.0 mm in thickness with ulceration | | T4a = Melanomas >4.0 mm in thickness without ulceration | | T4b = Melanomas >4.0 mm in thickness with ulceration | | Any N | NX = Regional lymph nodes cannot be assessed (e.g., previously removed for another reason) | | N1a = 1 regional lymph node metastasis with micrometastasisc | | N1b = 1 regional lymph node metastasis with macrometastasisd | | N2a = 2-3 regional lymph node metastases with micrometastasisc | | N2b = 2-3 regional lymph node metastases with macrometastasisd | | N2c = In transit met(s)/satellite(s) without metastatic lymph nodes | | N3 = ≥4 regional lymph node metastases; or matted nodes; or in transit met(s)/satellite(s) with metastatic lymph node(s) | | M1 | M1a = Metastases to skin, subcutaneous, or distant lymph nodes and normal serum LDH | | M1b = Metastases to lung and normal serum LDH | | M1c = Metastases to all other visceral sites and normal serum LDH; or distant metastases to any site and elevated serum LDH | | LDH = Lactate dehydrogenase; M = distant metastasis; N = regional lymph nodes; T = primary tumor. | | Adapted with permission from AJCC: Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44. | | a Clinical staging includes microstaging of the primary melanoma and clinical and/or radiologic evaluation for metastases. By convention, it should be used after complete excision of the primary melanoma with clinical assessment for regional and distant metastases. | | b Pathologic staging includes microstaging of the primary melanoma and pathologic information about the regional lymph nodes after partial or complete lymphadenectomy. Pathologic stage 0 or stage IA patients are the exception; they do not require pathologic evaluation of their lymph nodes. | | c Micrometastases are diagnosed after sentinel lymph node biopsy and complete lymphadenectomy (if performed). | | d Macrometastases are defined as clinically detectable nodal metastases confirmed by therapeutic lymphadenectomy or when nodal metastasis exhibits gross extracapsular extension. | References:
-
Melanoma of the skin. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 325-44.
Treatment Option Overview for MelanomaTable 8. Standard Treatment Options for Melanoma| Stage (TNM Staging Criteria) | Standard Treatment Optionsa |
|---|
| a Clinical trials are an important option for patients with all stages of melanoma because advances in understanding the aberrant molecular and biologic pathways have led to rapid drug development. Standard treatment options are available in many clinical trials. Information about ongoing clinical trials is available from theNCI website. | | Stage 0 melanoma | Excision | | Stage I melanoma | Excision+/− lymph node management | | Stage II melanoma | Excision+/− lymph node management | | Resectable Stage III melanoma | Excision+/− lymph node management | | Adjuvant therapyand immunotherapy | | Unresectable Stage III, Stage IV, and Recurrent melanoma | Intralesional therapy | | Immunotherapy | | Signal-transduction inhibitors | | Chemotherapy | | Palliative local therapy | Excision Surgical excision remains the primary modality for treating melanoma. Cutaneous melanomas that have not spread beyond the site at which they developed are highly curable. The treatment for localized melanoma is surgical excision with margins proportional to the microstage of the primary lesion. Lymph node management Sentinel lymph node biopsy (SLNB) Lymphatic mapping and SLNB can be considered to assess the presence of occult metastasis in the regional lymph nodes of patients with primary tumors larger than 1 to 4 mm, potentially identifying individuals who may be spared the morbidity of regional lymph node dissections and individuals who may benefit from adjuvant therapy.[1,2,3,4,5,6] To ensure accurate identification of the sentinel lymph node (SLN), lymphatic mapping and removal of the SLN should precede wide excision of the primary melanoma. Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[1,6,7,8,9,10,11] If metastatic melanoma is detected, a complete regional lymphadenectomy can be performed in a second procedure. Complete lymph node dissection (CLND) Patients can be considered for CLND if the sentinel node(s) is microscopically or macroscopically positive for regional control or considered for entry into the Multicenter Selective Lymphadenectomy Trial II (NCT00297895) to determine whether CLND affects survival. SLNB should be performed before wide excision of the primary melanoma to ensure accurate lymphatic mapping. Adjuvant Therapy High-dose interferon alpha-2b was approved by the U.S. Food and Drug Administration (FDA) in 1995 for the adjuvant treatment of patients with melanoma who have undergone a complete surgical resection but who are considered to be at a high risk of relapse (stages IIB, IIC, and III). However, prospective, randomized, multicenter treatment trials have demonstrated that high-dose interferon alpha-2b and pegylated interferon improve relapse-free survival but do not improve overall survival (OS). Therapies that have improved OS in patients with recurrent or metastatic disease are now being tested as adjuvant therapy in clinical trials, including NCT01274338, NCT01667419, and NCT01682083. Limb Perfusion A completed, multicenter, phase III randomized trial (SWOG-8593) of patients with high-risk primary stage I limb melanoma did not show a disease-free survival or OS benefit from isolated limb perfusion with melphalan, when compared with surgery alone.[5] Systematic Treatment for Unresectable Stage III, Stage IV, and Recurrent Disease Although melanoma that has spread to distant sites is rarely curable, treatment options are rapidly expanding. Two approaches-checkpoint inhibition and targeting the mitogen-activated protein kinase pathway-have demonstrated improvement in OS in randomized trials in comparison to dacarbazine (DTIC). Although none appear to be curative when used as single agents, early data of combinations are promising. Given the rapid development of new agents and combinations, patients and their physicians are encouraged to consider treatment in a clinical trial for initial treatment and at the time of progression. Immunotherapy Checkpoint inhibitors Three checkpoint inhibitors-pembrolizumab, nivolumab, and ipilimumab-are now approved by the FDA. Each has demonstrated the ability to impact OS against different comparators in unresectable or advanced disease. (Refer to the Pembrolizumab, the Nivolumab, and the Ipilimumab sections in the Unresectable Stage III, Stage IV, and Recurrent Melanoma Treatment section of this summary for more information.) Multiple phase III trials are in progress to determine optimal sequencing of immunotherapies, immunotherapy with targeted therapy, and whether combinations of immunotherapies or immunotherapy plus targeted therapy are superior for increasing OS. Interleukin-2 (IL-2) IL-2 was approved by the FDA in 1998 on the basis of durable complete response (CR) rates in a minority of patients (6%-7%) with previously treated metastatic melanoma in eight phase I and II studies. Phase III trials comparing high-dose IL-2 with other treatments and providing an assessment of relative impact on OS have not been conducted. Signal-transduction inhibitors Studies to date indicate that both BRAF and MEK inhibitors can significantly impact the natural history of melanoma, although they do not appear to be curative as single agents. BRAFinhibitors Vemurafenib Vemurafenib, approved by the FDA in 2011, has demonstrated an improvement in progression-free survival (PFS) and OS in patients with unresectable or advanced disease. Vemurafenib is an orally available, small-molecule, selective BRAF V600E kinase inhibitor, and its indication is limited to patients with a demonstrated BRAF V600E mutation by an FDA-approved test.[11] Dabrafenib Dabrafenib, an orally available, small-molecule, selective BRAF inhibitor that was approved by the FDA in 2013, showed improvement in PFS when compared with DTIC in an international, multicenter trial (BREAK-3 [NCT01227889]). MEKinhibitors Trametinib Trametinib is an orally available, small-molecule, selective inhibitor of MEK1 and MEK2 that was approved by the FDA in 2013 for patients with unresectable or metastatic melanoma with BRAF V600E or K mutations. Trametinib demonstrated improved PFS over DTIC. Cobimetinib Cobimetinib is an orally available, small-molecule, selective MEK inhibitor that was approved by the FDA in 2015 for use in combination with the BRAF inhibitor vemurafenib. (Refer to the Combination signal-transduction inhibitor therapy section of this summary for more information.) c-KIT inhibitors Early data suggest that mucosal or acral melanomas with activating mutations or amplifications in c-KIT may be sensitive to a variety of c-KIT inhibitors.[12,13,14] Phase II and phase III trials are available for patients with unresectable stage III or stage IV melanoma harboring the c-KIT mutation. Combination signal-transduction inhibitor therapy In 2014, the combination of dabrafenib and trametinib received accelerated approval from the FDA for patients with unresectable or metastatic melanomas that carry the BRAF V600E or V600K mutation. The combination demonstrated improved durable response rates over single-agent dabrafenib. Full approval is pending completion of ongoing clinical trials and demonstration of clinical benefit on OS. In 2015, the combination of vemurafenib and cobimetinib was also approved by the FDA for patients with unresectable or metastatic melanomas that carry the BRAF V600E or V600 K mutation. Published phase III data support improved PFS of another combination of BRAF and MEK inhibitors versus BRAF inhibitor plus placebo; dabrafenib plus trametinib compared with dabrafenib plus placebo. OS data are immature. Chemotherapy DTIC DTIC was approved in 1970 on the basis of overall response rates. Phase III trials indicate an overall response rate of 10% to 20%, with rare complete responses observed. An impact on OS has not been demonstrated in randomized trials.[15,16,17,18] When used as a control arm for recent registration trials of ipilimumab and vemurafenib in previously untreated patients with metastatic melanoma, DTIC was shown to be inferior for OS. Temozolomide Temozolomide, an oral alkylating agent, appeared to be similar to intravenous DTIC in a randomized phase III trial with a primary endpoint of OS; however, because the trial was designed to demonstrate the superiority of temozolomide, which was not achieved, the trial was left with a sample size that was inadequate to provide statistical proof of noninferiority.[16] Palliative local therapy Melanoma metastatic to distant, lymph node-bearing areas may be palliated by regional lymphadenectomy. Isolated metastases to the lung, gastrointestinal tract, bone, or sometimes the brain may be palliated by resection, with occasional long-term survival.[19,20,21] References:
-
Shen P, Wanek LA, Morton DL: Is adjuvant radiotherapy necessary after positive lymph node dissection in head and neck melanomas? Ann Surg Oncol 7 (8): 554-9; discussion 560-1, 2000.
-
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998.
-
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000.
-
Cascinelli N, Morabito A, Santinami M, et al.: Immediate or delayed dissection of regional nodes in patients with melanoma of the trunk: a randomised trial. WHO Melanoma Programme. Lancet 351 (9105): 793-6, 1998.
-
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998.
-
Wong SL, Balch CM, Hurley P, et al.: Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. J Clin Oncol 30 (23): 2912-8, 2012.
-
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996.
-
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000.
-
Eggermont AM, Suciu S, Santinami M, et al.: Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet 372 (9633): 117-26, 2008.
-
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study--United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004.
-
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011.
-
Hodi FS, Friedlander P, Corless CL, et al.: Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046-51, 2008.
-
Guo J, Si L, Kong Y, et al.: Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29 (21): 2904-9, 2011.
-
Carvajal RD, Antonescu CR, Wolchok JD, et al.: KIT as a therapeutic target in metastatic melanoma. JAMA 305 (22): 2327-34, 2011.
-
Chapman PB, Einhorn LH, Meyers ML, et al.: Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 17 (9): 2745-51, 1999.
-
Middleton MR, Grob JJ, Aaronson N, et al.: Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18 (1): 158-66, 2000.
-
Avril MF, Aamdal S, Grob JJ, et al.: Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol 22 (6): 1118-25, 2004.
-
Robert C, Thomas L, Bondarenko I, et al.: Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364 (26): 2517-26, 2011.
-
Leo F, Cagini L, Rocmans P, et al.: Lung metastases from melanoma: when is surgical treatment warranted? Br J Cancer 83 (5): 569-72, 2000.
-
Ollila DW, Hsueh EC, Stern SL, et al.: Metastasectomy for recurrent stage IV melanoma. J Surg Oncol 71 (4): 209-13, 1999.
-
Gutman H, Hess KR, Kokotsakis JA, et al.: Surgery for abdominal metastases of cutaneous melanoma. World J Surg 25 (6): 750-8, 2001.
Stage 0 Melanoma TreatmentStandard Treatment Options for Stage 0 Melanoma Standard treatment options for stage 0 melanoma include the following: - Excision.
Excision Patients with stage 0 disease may be treated by excision with minimal, but microscopically free, margins. Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with stage 0 melanoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. Stage I Melanoma TreatmentStandard Treatment Options for Stage I Melanoma Standard treatment options for stage I melanoma include the following: - Excision with or without lymph node management.
Excision Evidence suggests that lesions no thicker than 2 mm may be treated conservatively with radial excision margins of 1 cm. Depending on the location of the melanoma, most patients can now have the excision performed on an outpatient basis. Evidence (excision): - A randomized trial compared narrow margins (1 cm) with wide margins (≥3 cm) in patients with melanomas no thicker than 2 mm.[1,2][Level of evidence: 1iiA]
- No difference was observed between the two groups in the development of metastatic disease, disease-free survival (DFS), or overall survival (OS).
- Two other randomized trials compared 2-cm margins with wider margins (4 cm or 5 cm).[3,4][Level of evidence:1iiA]
- No statistically significant difference in local recurrence, distant metastasis, or OS was found; the median follow-up was at least 10 years for both trials.
- In the Intergroup Melanoma Surgical Trial, the reduction in margins from 4 cm to 2 cm was associated with both of the following:[5][Level of evidence: 1iiA]
- A statistically significant reduction in the need for skin grafting (from 46% to 11%; P < .001).
- A reduction in the length of hospital stay.
- A multicenter, phase III randomized trial (SWOG-8593) of patients with high-risk stage I primary limb melanoma did not show a DFS or OS benefit from isolated limb perfusion with melphalan, when compared with surgery alone.[6,7]
Lymph node management Elective regional lymph node dissection is of no proven benefit for patients with stage I melanoma.[8] Lymphatic mapping and sentinel lymph node biopsy (SLNB) for patients who have tumors of intermediate thickness and/or ulcerated tumors may identify individuals with occult nodal disease. These patients may benefit from regional lymphadenectomy and adjuvant therapy.[6,9,10,11] Evidence (immediate lymphadenectomy vs. observation with delayed lymphadenectomy): - The International Multicenter Selective Lymphadenectomy Trial (MSLT-1 [JWCI-MORD-MSLT-1193]) included 1,269 patients with intermediate-thickness (defined as 1.2 mm-3.5 mm in this study) primary melanomas.[12][Level of evidence: 1iiB]
- There was no melanoma-specific survival advantage (primary endpoint) for patients randomly assigned to undergo wide excision plus SLNB, followed by immediate complete lymphadenectomy for node positivity versus nodal observation and delayed lymphadenectomy for subsequent nodal recurrence at a median of 59.8 months.
- This trial was not designed to detect a difference in the impact of lymphadenectomy in patients with microscopic lymph node involvement.
- The Sunbelt Melanoma Trial (UAB-9735 [NCT00004196]) was a phase III trial to determine the effects of lymphadenectomy with or without adjuvant high-dose interferon alpha-2b versus observation on DFS and OS in patients with submicroscopic sentinel lymph node (SLN) metastasis detected only by the polymerase chain reaction assay (i.e., SLN negative by histology and immunohistochemistry).
- No survival data have been reported from this study.
Treatment Options Under Clinical Evaluation for Stage I Melanoma Treatment options under clinical evaluation for patients with stage I melanoma include the following: - Clinical trials evaluating new techniques to detect submicroscopic SLN metastasis. Because of the higher rate of treatment failure in the subset of clinical stage I patients with occult nodal disease, clinical trials have evaluated new techniques to detect submicroscopic SLN metastasis to identify patients who may benefit from regional lymphadenectomy with or without adjuvant therapy.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with stage I melanoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Veronesi U, Cascinelli N: Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. Arch Surg 126 (4): 438-41, 1991.
-
Veronesi U, Cascinelli N, Adamus J, et al.: Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med 318 (18): 1159-62, 1988.
-
Cohn-Cedermark G, Rutqvist LE, Andersson R, et al.: Long term results of a randomized study by the Swedish Melanoma Study Group on 2-cm versus 5-cm resection margins for patients with cutaneous melanoma with a tumor thickness of 0.8-2.0 mm. Cancer 89 (7): 1495-501, 2000.
-
Balch CM, Soong SJ, Smith T, et al.: Long-term results of a prospective surgical trial comparing 2 cm vs. 4 cm excision margins for 740 patients with 1-4 mm melanomas. Ann Surg Oncol 8 (2): 101-8, 2001.
-
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993.
-
Essner R, Conforti A, Kelley MC, et al.: Efficacy of lymphatic mapping, sentinel lymphadenectomy, and selective complete lymph node dissection as a therapeutic procedure for early-stage melanoma. Ann Surg Oncol 6 (5): 442-9, 1999 Jul-Aug.
-
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998.
-
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998.
-
Gershenwald JE, Thompson W, Mansfield PF, et al.: Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol 17 (3): 976-83, 1999.
-
Mraz-Gernhard S, Sagebiel RW, Kashani-Sabet M, et al.: Prediction of sentinel lymph node micrometastasis by histological features in primary cutaneous malignant melanoma. Arch Dermatol 134 (8): 983-7, 1998.
-
Morton DL, Thompson JF, Cochran AJ, et al.: Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355 (13): 1307-17, 2006.
-
Morton DL, Thompson JF, Cochran AJ, et al.: Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med 370 (7): 599-609, 2014.
Stage II Melanoma TreatmentStandard Treatment Options for Stage II Melanoma Standard treatment options for stage II melanoma include the following: - Excision with or without lymph node management.
Excision For melanomas with a thickness between 2 mm and 4 mm, surgical margins need to be 2 cm to 3 cm or smaller. Few data are available to guide treatment in patients with melanomas thicker than 4 mm; however, most guidelines recommend margins of 3 cm whenever anatomically possible. Depending on the location of the melanoma, most patients can have the excision performed on an outpatient basis. Evidence (excision): - The Intergroup Melanoma Surgical Trial Task 2b compared 2-cm versus 4-cm margins for patients with melanomas that were 1 mm to 4 mm thick.[1]
- With a median follow-up of more than 10 years, no significant difference in local recurrence or survival was observed between the two groups.
- The reduction in margins from 4 cm to 2 cm was associated with the following:
- A statistically significant reduction in the need for skin grafting (from 46% to 11%; P < .001).
- A reduction in the length of the hospital stay.
- A study conducted in the United Kingdom randomly assigned patients with melanomas thicker than 2 mm to undergo excision with either 1-cm or 3-cm margins.[2]
- Patients treated with excision with 1-cm margins had higher rates of local regional recurrence (hazard ratio [HR], 1.26; 95% confidence interval [CI], 1.00-1.59; P = .05).
- No difference in survival was seen (HR, 1.24; 95% CI, 0.96-1.61; P = .1).
- This study suggests that 1-cm margins may not be adequate for patients with melanomas thicker than 2 mm.
Lymph Node Management Lymphatic mapping and sentinel lymph node biopsy (SLNB) Lymphatic mapping and SLNB have been used to assess the presence of occult metastasis in the regional lymph nodes of patients with stage II disease, potentially identifying individuals who may be spared the morbidity of regional lymph node dissections (LNDs) and individuals who may benefit from adjuvant therapy.[3,4,5,6,7] To ensure accurate identification of the sentinel lymph node, lymphatic mapping and removal of the SLN should precede wide excision of the primary melanoma. With the use of a vital blue dye and a radiopharmaceutical agent injected at the site of the primary tumor, the first lymph node in the lymphatic basin that drains the lesion can be identified, removed, and examined microscopically. Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[3,8,9,10,11,12] If metastatic melanoma is detected, a complete regional lymphadenectomy can be performed in a second procedure. Regional lymphadenectomy No published data on the clinical significance of micrometastatic melanoma in regional lymph nodes are available from prospective trials. Some evidence suggests that for patients with tumors of intermediate thickness and occult metastasis, survival is better among patients who undergo immediate regional lymphadenectomy than it is among those who delay lymphadenectomy until the clinical appearance of nodal metastasis.[13] This finding should be viewed with caution because it arose from a post hoc subset analysis of data from a randomized trial. Evidence (regional lymphadenectomy): - The International Multicenter Selective Lymphadenectomy Trial (MSLT-1 [JWCI-MORD-MSLT-1193]) included 1,269 patients with intermediate-thickness (defined as 1.2 mm-3.5 mm in this study) primary melanomas.[14][Level of evidence: 1iiB]
- There was no melanoma-specific survival advantage (primary endpoint) for patients randomly assigned to undergo wide excision plus SLNB, followed by immediate complete lymphadenectomy for node positivity versus nodal observation and delayed lymphadenectomy for subsequent nodal recurrence at a median of 59.8 months.
- This trial was not designed to detect a difference in the impact of lymphadenectomy in patients with microscopic lymph node involvement.
- Three other prospective randomized trials have failed to show a survival benefit for prophylactic regional LNDs.[15,16,17]
Adjuvant therapy High-dose interferon High-dose interferon alpha-2b was approved in 1995 for the adjuvant treatment of patients with melanoma who have undergone a complete surgical resection but are considered to be at a high risk of relapse. Evidence was based on a significantly improved relapse-free survival (RFS) and marginally improved overall survival (OS) that were seen in EST-1684. Subsequent large, randomized trials have not been able to reproduce a benefit in OS. Ongoing trials are testing therapies that have demonstrated improved OS in patients with stage IV disease. Clinicians should be aware that the high-dose regimens have significant toxic effects. Evidence (high-dose interferon alpha-2b): - A multicenter, randomized, controlled study (EST-1684) compared a high-dose regimen of interferon alpha-2b (20 mU/m2 of body surface per day given intravenously 5 days a week for 4 weeks, then 10 mU/m2 of body surface per day given subcutaneously 3 times a week for 48 weeks) with observation.[8][Level of evidence: 1iiA]
- This study included 287 patients at high risk of recurrence after potentially curative surgery for melanoma (patients with melanomas thicker than 4 mm without involved lymph nodes or patients with melanomas of any thickness with positive lymph nodes).
- Patients who had recurrent melanoma involving only the regional lymph nodes were also eligible.
- At a median follow-up of 7 years, this trial demonstrated a significant prolongation of RFS (P = .002) and OS (P = .024) for patients who received high-dose interferon.
- The median OS for patients who received the high-dose regimen of interferon alpha-2b was 3.8 years, compared with 2.8 years for those in the observation group.
- A subset analysis of the stage II patients failed to show any RFS or OS benefit from high-dose interferon. Because the number of stage II patients was small in this subset analysis, it is difficult to draw meaningful conclusions from this study for this specific group.
- A multicenter, randomized, controlled study (EST-1690) conducted by the same investigators compared the same high-dose interferon alpha regimen with either a low-dose regimen of interferon alpha-2b (3 mU/m2 of body surface per day given subcutaneously three times per week for 104 weeks) or observation. The stage entry criteria for this trial included patients with stage II and III melanoma. This three-arm trial enrolled 642 patients.[9][Level of evidence: 1iiA]
- At a median follow-up of 52 months, a statistically significant RFS advantage was shown for all patients who received high-dose interferon (including the clinical stage II patients) when compared with the observation group (P = .03).
- No statistically significant RFS advantage was seen for patients who received low-dose interferon when compared with the observation group.
- The 5-year estimated RFS rate was 44% for the high-dose interferon group, 40% for the low-dose interferon group, and 35% for the observation group.
- Neither high-dose nor low-dose interferon yielded an OS benefit when compared with observation (HR, 1.0; P = .995).
Treatment Options Under Clinical Evaluation for Stage II Melanoma Postsurgical adjuvant treatment (e.g., with interferons) has not been shown to affect survival. Treatment options under clinical evaluation for patients with stage II melanoma include the following: - Clinical trials are testing therapies of postsurgical adjuvant treatment that have improved OS in patients with stage IV disease, including NCT01274338, NCT01667419, and NCT01682083. Postsurgical adjuvant treatment (e.g., with interferons) has not been shown to affect survival; therefore, clinical trials are an important therapeutic option for patients at high risk for relapse.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with stage II melanoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993.
-
Thomas JM, Newton-Bishop J, A'Hern R, et al.: Excision margins in high-risk malignant melanoma. N Engl J Med 350 (8): 757-66, 2004.
-
Gershenwald JE, Thompson W, Mansfield PF, et al.: Multi-institutional melanoma lymphatic mapping experience: the prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J Clin Oncol 17 (3): 976-83, 1999.
-
McMasters KM, Reintgen DS, Ross MI, et al.: Sentinel lymph node biopsy for melanoma: controversy despite widespread agreement. J Clin Oncol 19 (11): 2851-5, 2001.
-
Cherpelis BS, Haddad F, Messina J, et al.: Sentinel lymph node micrometastasis and other histologic factors that predict outcome in patients with thicker melanomas. J Am Acad Dermatol 44 (5): 762-6, 2001.
-
Essner R: The role of lymphoscintigraphy and sentinel node mapping in assessing patient risk in melanoma. Semin Oncol 24 (1 Suppl 4): S8-10, 1997.
-
Chan AD, Morton DL: Sentinel node detection in malignant melanoma. Recent Results Cancer Res 157: 161-77, 2000.
-
Morton DL, Wen DR, Wong JH, et al.: Technical details of intraoperative lymphatic mapping for early stage melanoma. Arch Surg 127 (4): 392-9, 1992.
-
Reintgen D, Cruse CW, Wells K, et al.: The orderly progression of melanoma nodal metastases. Ann Surg 220 (6): 759-67, 1994.
-
Thompson JF, McCarthy WH, Bosch CM, et al.: Sentinel lymph node status as an indicator of the presence of metastatic melanoma in regional lymph nodes. Melanoma Res 5 (4): 255-60, 1995.
-
Uren RF, Howman-Giles R, Thompson JF, et al.: Lymphoscintigraphy to identify sentinel lymph nodes in patients with melanoma. Melanoma Res 4 (6): 395-9, 1994.
-
Bostick P, Essner R, Glass E, et al.: Comparison of blue dye and probe-assisted intraoperative lymphatic mapping in melanoma to identify sentinel nodes in 100 lymphatic basins. Arch Surg 134 (1): 43-9, 1999.
-
Cascinelli N, Morabito A, Santinami M, et al.: Immediate or delayed dissection of regional nodes in patients with melanoma of the trunk: a randomised trial. WHO Melanoma Programme. Lancet 351 (9105): 793-6, 1998.
-
Morton DL, Thompson JF, Cochran AJ, et al.: Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355 (13): 1307-17, 2006.
-
Veronesi U, Adamus J, Bandiera DC, et al.: Delayed regional lymph node dissection in stage I melanoma of the skin of the lower extremities. Cancer 49 (11): 2420-30, 1982.
-
Sim FH, Taylor WF, Ivins JC, et al.: A prospective randomized study of the efficacy of routine elective lymphadenectomy in management of malignant melanoma. Preliminary results. Cancer 41 (3): 948-56, 1978.
-
Balch CM, Soong SJ, Bartolucci AA, et al.: Efficacy of an elective regional lymph node dissection of 1 to 4 mm thick melanomas for patients 60 years of age and younger. Ann Surg 224 (3): 255-63; discussion 263-6, 1996.
Resectable Stage III Melanoma TreatmentStandard Treatment Options for Resectable Stage III Melanoma Standard treatment options for resectable stage III melanoma include the following: - Excision with or without lymph node management.
- Adjuvant therapy and immunotherapy.
Excision The primary tumor may be treated with wide local excision with 1-cm to 3-cm margins, depending on tumor thickness and location.[1,2,3,4,5,6,7] Skin grafting may be necessary to close the resulting defect. Lymph node management Sentinel lymph node biopsy (SLNB) Lymphatic mapping and SLNB can be considered to assess the presence of occult metastases in the regional lymph nodes of patients with primary tumors larger than 1 mm to 4 mm, potentially identifying individuals who may be spared the morbidity of regional lymph node dissections and individuals who may benefit from adjuvant therapy.[3,8,9,10,11,12] To ensure accurate identification of the sentinel lymph node (SNL), lymphatic mapping and removal of the SLN should precede wide excision of the primary melanoma. Multiple studies have demonstrated the diagnostic accuracy of SLNB, with false-negative rates of 0% to 2%.[8,12,13,14,15,16,17] If metastatic melanoma is detected, a complete regional lymphadenectomy can be performed in a second procedure. Complete lymph node dissection (CLND) Patients can be considered for CLND if the sentinel node(s) is microscopically or macroscopically positive for regional control or considered for entry into the Multicenter Selective Lymphadenectomy Trial II to determine whether CLND affects survival. SLNB should be performed prior to wide excision of the primary melanoma to ensure accurate lymphatic mapping. Adjuvant Therapy Prospective, randomized, multicenter treatment trials have demonstrated that ipilimumab, high-dose interferon alpha-2b, and pegylated interferon can improve relapse-free survival (RFS). Data are immature to know whether ipilimumab can impact overall survival (OS), but trials have shown that interferon alone does not. A number of single drugs or combinations of drugs, which have demonstrated improved OS in patients with advanced disease, are being tested in clinical trials of adjuvant therapy in patients at high risk for relapse after surgical resection of tumors. These trials include immunotherapy and therapies targeted to specific mutations. Examples include NCT01274338, NCT01667419, NCT01682083, and NCT02362594, among others. Immunotherapy Ipilimumab Evidence (ipilimumab): - In a multinational, randomized, double-blind trial, patients with stage III melanoma, who had complete resection, were randomly assigned (1:1) to receive ipilimumab or placebo.[18][Level of evidence: 1iiDii] Patients with lymph node metastasis larger than 1 mm, in-transit metastasis, resection occurring more than 12 weeks before randomization, autoimmune disease, previous or concurrent immunosuppressive therapy, previous systemic therapy for melanoma, and an Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) score of greater than 1 were excluded. The ipilimumab dose was 10 mg/kg every 3 weeks for four doses, then every 3 months for up to 3 years. The primary endpoint was RFS, defined as recurrence or death (regardless of cause), whichever came first, as assessed by an independent review committee.
- A total of 951 patients were enrolled (475 patients to the ipilimumab arm and 476 patients to the placebo arm). The median age was 51 years, and 94% of the patients had a PS of 0.
- At a median follow-up of 2.7 years, there were 528 RFS events: 234 in the ipilimumab group (49%; 220 recurrences, 14 deaths) and 294 in the placebo group (62%; 289 recurrences, 5 deaths). Median RFS was 26 months for the ipilimumab group (95% CI, 19-39) versus 17 months for the placebo group (95% CI, 13-22). The hazard ratio (HR) was 0.75 (95% CI, 0.64-0.90); P < .002. The effect of ipilimumab was consistent across subgroups.
- It is not known whether adjuvant ipilimumab has an impact on OS. Toxicities may have led to inadvertent blinding. The final analysis for OS will be reported later.
- Ipilimumab was discontinued for adverse events (AEs) in 52% of the patients. Patients received ipilimumab for a median of four doses; 36% of patients in the ipilimumab group stayed on treatment for more than 6 months, and 26% stayed on treatment for more than 1 year. Five patients died from drug-related events: three secondary to colitis, one with myocarditis, and one of multiorgan failure with Guillain-Barré syndrome. The most common AEs were gastrointestinal, hepatic, and endocrine in nature and included rash, fatigue, and headache.
The optimal dose in the adjuvant setting is unknown. The recommended dose for patients with metastatic disease is 3 mg/kg. An intergroup trial, NCT01274338, is open to enrollment and designed to test whether high- versus low-dose ipilimumab or interferon is optimal. Interferon alpha-2b Evidence (high-dose interferon alpha): - A multicenter, randomized, controlled study (EST-1690) compared a high-dose interferon alpha regimen with either a low-dose regimen of interferon alpha-2b (3 mU/m2 of body surface per day given subcutaneously three times per week for 104 weeks) or observation. The stage entry criteria for this trial included patients with stage II and III melanoma. This three-arm trial enrolled 642 patients.[14][Level of evidence: 1iiA]
- At a median follow-up of 52 months, a statistically significant RFS advantage was shown for all patients who received high-dose interferon (including the clinical stage II patients) when compared with the observation group (P = .03).
- No statistically significant RFS advantage was seen for patients who received low-dose interferon when compared with the observation group.
- The 5-year estimated RFS rate was 44% for the high-dose interferon group, 40% for the low-dose interferon group, and 35% for the observation group.
- Neither high-dose nor low-dose interferon yielded an OS benefit when compared with observation (HR, 1.0; P = .995).
- Pooled analyses (EST-1684 and EST-1690) of the high-dose arms versus the observation arms suggest that treatment confers a significant RFS advantage but not a significant benefit for survival.
- A randomized, multicenter, national trial, ECOG-1697 [NCT00003641], evaluated high-dose intravenous interferon for a short duration (1 month) versus observation in patients with node-negative melanoma at least 2 mm thick or with any thickness and positive sentinel nodes. This trial was closed at interim analysis because of the lack of benefit from treatment with interferon.
- In 2011, pegylated interferon alpha-2b, which is characterized by a longer half-life and can be administered subcutaneously, was approved by the U.S. Food and Drug Administration for the adjuvant treatment of melanoma with microscopic or gross nodal involvement within 84 days of complete surgical resection, including complete lymphadenectomy.
Approval of pegylated interferon alpha-2b was based on EORTC-18991 [NCT00006249], which randomly assigned 1,256 patients with resected stage III melanoma to observation or weekly subcutaneous pegylated interferon alpha-2b for up to 5 years.[15][Level of evidence: 1iiDii]
- RFS, as determined by an independent review committee, was improved for patients receiving interferon (34.8 months vs. 25.5 months in the observation arm; HR, 0.82; 95% CI, 0.71-0.96; P = .011).
- No difference in median OS between the arms was observed (HR, 0.98; 95% CI, 0.82-1.16).
- One-third of the patients receiving pegylated interferon discontinued treatment because of toxicity.
Treatment Options Under Clinical Evaluation for Resectable Stage III Melanoma Treatment options under clinical evaluation for patients with resectable stage III melanoma include the following: - Trials of adjuvant therapies that have shown improvement in OS in metastatic disease.
- Trials of adjuvant therapies that target a known mutation, e.g., c-KIT.
- Intralesional therapies.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with stage III melanoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Veronesi U, Cascinelli N: Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. Arch Surg 126 (4): 438-41, 1991.
-
Veronesi U, Cascinelli N, Adamus J, et al.: Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. N Engl J Med 318 (18): 1159-62, 1988.
-
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000.
-
Cohn-Cedermark G, Rutqvist LE, Andersson R, et al.: Long term results of a randomized study by the Swedish Melanoma Study Group on 2-cm versus 5-cm resection margins for patients with cutaneous melanoma with a tumor thickness of 0.8-2.0 mm. Cancer 89 (7): 1495-501, 2000.
-
Balch CM, Soong SJ, Smith T, et al.: Long-term results of a prospective surgical trial comparing 2 cm vs. 4 cm excision margins for 740 patients with 1-4 mm melanomas. Ann Surg Oncol 8 (2): 101-8, 2001.
-
Heaton KM, Sussman JJ, Gershenwald JE, et al.: Surgical margins and prognostic factors in patients with thick (>4mm) primary melanoma. Ann Surg Oncol 5 (4): 322-8, 1998.
-
Balch CM, Urist MM, Karakousis CP, et al.: Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. Ann Surg 218 (3): 262-7; discussion 267-9, 1993.
-
Shen P, Wanek LA, Morton DL: Is adjuvant radiotherapy necessary after positive lymph node dissection in head and neck melanomas? Ann Surg Oncol 7 (8): 554-9; discussion 560-1, 2000.
-
Hochwald SN, Coit DG: Role of elective lymph node dissection in melanoma. Semin Surg Oncol 14 (4): 276-82, 1998.
-
Cascinelli N, Morabito A, Santinami M, et al.: Immediate or delayed dissection of regional nodes in patients with melanoma of the trunk: a randomised trial. WHO Melanoma Programme. Lancet 351 (9105): 793-6, 1998.
-
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998.
-
Wong SL, Balch CM, Hurley P, et al.: Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. J Clin Oncol 30 (23): 2912-8, 2012.
-
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996.
-
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000.
-
Eggermont AM, Suciu S, Santinami M, et al.: Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet 372 (9633): 117-26, 2008.
-
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study--United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004.
-
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011.
-
Eggermont AM, Chiarion-Sileni V, Grob JJ, et al.: Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 16 (5): 522-30, 2015.
Unresectable Stage III, Stage IV, and Recurrent Melanoma TreatmentTreatment Options for Unresectable Stage III, Stage IV, and Recurrent Melanoma Treatment options for unresectable stage III, stage IV, and recurrent melanoma include the following: - Intralesional therapy.
- Immunotherapy.
- Checkpoint inhibitors.
- Anti-PD-1 (programmed cell death-1) and PD-L1 (programmed death ligand 1).
- Anti-CTLA-4 (cytotoxic T-lymphocyte antigen-4): ipilimumab.
- High-dose interleukin-2 (IL-2).
- Dual immunomodulation.
- Dual checkpoint inhibition.
- Signal-transduction inhibitors.
- BRAF (V-raf murine sarcoma viral oncogene homolog B1) inhibitors (for patients who test positive for the BRAF V600 mutation).
- MEK inhibitors.
- KIT inhibitors.
- Combination therapy with signal-transduction inhibitors.
- BRAF plus MEK inhibitors.
- Dabrafenib plus trametinib.
- Vemurafenib plus cobimetinib.
- Multikinase inhibitors.
- Chemotherapy.
- Palliative local therapy.
Two approaches-checkpoint inhibition and targeting the mitogen-activated protein kinase (MAPK) pathway-have demonstrated improvement in overall survival (OS) in randomized trials versus the use of dacarbazine (DTIC) or in comparison to DTIC. Given the rapid development of new agents and combinations, patients and their physicians are encouraged to consider treatment in a clinical trial for initial treatment and at the time of progression. Intralesional therapy Talimogene laherparepvec (T-VEC) T-VEC is a genetically modified, herpes simplex virus type 1 (HSV1) oncolytic therapy approved for local intralesional injection into unresectable cutaneous, subcutaneous, and nodal lesions in patients with melanoma that recurs after initial surgery. T-VEC is designed to replicate within tumors, causing lysis, and to produce granulocyte-macrophage colony-stimulating factor (GM-CSF). Release of antigens together with virally derived GM-CSF may promote an antitumor immune response; however, the exact mechanism of action is unknown. The approval of T-VEC by the U.S. Food and Drug Administration (FDA) is based on data that demonstrated shrinkage of lesions; however, improvement of OS or an effect on visceral metastases or improvement in quality of life has not been shown. Evidence (T-VEC): - In a multinational, randomized, open-label trial (NCT00769704), 436 patients were randomly assigned 2:1 to intralesional T-VEC or subcutaneous GM-CSF for at least 6 months or until there were no more injectable lesions.[1][Level of evidence: 1iiDiv] Eligible patients had stage IIIB, IIIC, and IV melanoma with unresectable, bidimensionally measurable lesions. The primary endpoint was durable response rate (DRR) (complete response [CR] or partial response [PR] lasting for >6 months) as assessed by independent review. The study was stratified by site of first recurrence, presence of liver metastases, disease stage, and previous nonadjuvant systemic treatment.
- The median patient age was 63 years (range 22-94 years), 70% of patients had a baseline Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) score of 0, 30% had stage III disease, and 70% had stage IV disease (27% M1a; 21% M1b; and 22% M1c). Previous therapy for melanoma had been received by 53% of the patients.
- The first dose only was administered at 106 plaque-forming units (pfu)/mL to a maximum of 4 mL for all lesions combined. Subsequent doses were administered at 8 pfu/mL up to 4.0 mL for all injected lesions combined with the injected volume based on the size of the lesion. Injection into visceral lesions was not allowed.
- In patients treated with T-VEC, 16% (95% confidence interval [CI], 12.0-20.5) had a DRR versus 2% (95% CI, 0-4.5) in patients receiving GM-CSF. Subgroup analysis suggests that the differences in DRRs between T-VEC versus GM-CSF may be greater in earlier-stage disease and treatment-naïve disease. Patients with stage IIIB and IIIC had a 33% DRR with T-VEC versus 0% with GM-CSF; 16% versus 2%, respectively in patients with stage IV M1a disease; 3% versus 4%, respectively in patients with stage IV M1b disease; and, 8% versus 3%, respectively in patients with stage IV M1c disease. Patients treated with T-VEC or GM-CSF as first-line therapy had a DRR of 24% versus 0%; however, patients who received treatment as second-line therapy or greater had a DRR of 10% versus 4%.
- The median duration of exposure to T-VEC was 23 weeks (5.3 months) with 26 patients exposed for more than 1 year. The most common adverse events (AEs) in the T-VEC group were fatigue (50%), chills (49%), pyrexia (43%), nausea (36%), influenza-like illness (30%), and injection site pain. The rate of discontinuation resulting from toxicity to T-VEC was 4% versus 2% in the GM-CSF group. Of the ten deaths in patients treated with T-VEC, eight deaths were considered the result of PD-1, salmonella infection, and one myocardial infarction; none were considered related to therapy, based on findings of investigator.
Precautions: T-VEC is a live, attenuated herpes simplex virus and may cause life-threatening, disseminated herpetic infection. It is contraindicated in immunocompromised or pregnant patients. Healthcare providers and close contacts should avoid direct contact with injected lesions. Biohazard precautions for preparation, administration, and handling are provided in the label. Detailed prescribing information by treatment cycle and lesion size are provided in the FDA label. Immunotherapy Checkpoint inhibitors Anti-PD-1 and PD-L1 The PD-1 pathway is a key immunoinhibitory mediator of T-cell exhaustion. Blockade of this pathway can lead to T-cell activation, expansion, and enhanced effector functions. PD-1 has two ligands, PD-L1 and PD-L2 (Programmed Death-2 Ligand 2). Two anti-PD-1 antibodies, pembrolizumab and nivolumab, have gained accelerated approval from the FDA in 2014, based on DRRs in previously treated patients. Full approval will depend on a demonstration of improvement in progression-free survival (PFS), OS, or improved quality of life in randomized trials. Pembrolizumab Evidence (pembrolizumab): - Previously treated patients. A total of 173 patients with unresectable or metastatic melanoma with disease progression within 24 weeks of the last dose of ipilimumab and, if BRAF V600 mutation positive, previous treatment with a BRAF inhibitor, were randomly assigned to one of two doses of pembrolizumab-2 mg/kg or 10 mg/kg-every 3 weeks. The trial excluded patients with an autoimmune disease, a condition requiring immunosuppression, or a history of severe immune-related adverse events (irAEs) from treatment with ipilimumab.
- The median age was 61 years; 60% were male; 67% had an ECOG PS of 0, and 33% had an ECOG PS of 1. Eighteen percent of patients had tumors that were BRAF V600 mutation positive, 39% had an elevated lactate dehydrogenase (LDH), 64% had M1c disease, 9% had brain metastases, and 72% had undergone two or more therapies for advanced disease. The primary outcome measure was overall response rate (ORR) according to Response Evaluation Criteria In Solid Tumors (RECIST, version 1.1) criteria as assessed by blinded independent central review.[2][Level of evidence: 1iiDiv]
- The ORR determined by independent central review was 26% (95% CI, -14-13; P = .96) in the 2 mg/kg arm, consisting of one CR and 20 PRs in 81 patients. Median follow-up was 8 months, and all patients had a minimum of 6 months of follow-up. Among the 21 patients with an objective response, 18 had ongoing responses, ranging from 1.4+ months to 8.5+ months.
- Response rate in the 10 mg/kg arm was similar at 26%, consisting of 20 responses in 76 patients. Responses were seen in patients with and without BRAF V600 mutations.
- The approved dose was 2 mg/kg administered as an intravenous (IV) infusion for 30 minutes every 3 weeks.
Pembrolizumab was discontinued because of AEs in 7% of the patients treated with 2 mg/kg, with 3% considered drug-related AEs by the investigators. The most common AEs in the 2 mg/kg versus 10 mg/kg arms were: - Fatigue (33% vs. 37%).
- Pruritus (23% vs. 19%).
- Rash (18% vs. 18%).
Other common AEs included cough, nausea, decreased appetite, constipation, arthralgia, and diarrhea. The most frequent and serious AEs that occurred in more than 2% of a total of 411 patients treated with pembrolizumab included renal failure, dyspnea, pneumonia, and cellulitis. Additional clinically significant irAEs included pneumonitis, colitis, hypophysitis, hyperthyroidism, hypothyroidism, nephritis, and hepatitis. The FDA label provides recommendations for suspected irAEs, including withholding the drug and administering corticosteroids. - Previously untreated and treated patients. A multicenter, international trial (KEYNOTE 006 [NCT01866319]) randomly assigned 834 patients with metastatic melanoma in a 1:1:1 ratio to receive pembrolizumab (10 mg/kg IV every 2 weeks or every 3 weeks) or four cycles of ipilimumab (3 mg/kg every 3 weeks).[3] Patients were stratified by ECOG PS (0 vs. 1), line of therapy (first-line vs. second-line), and PD-L1 expression (positive vs. negative). The primary endpoints were PFS and OS.[3][Level of evidence: 1iiA]
Approximately 66% of patients had received no previous systemic therapy for advanced melanoma. BRAF V600 mutations were present in 36% of patients and of these, approximately 50% had received previous BRAF inhibitor treatments. The study did not enroll patients with BRAF V600 mutations with high LDH levels and symptomatic or rapidly progressive disease who had not received anti-BRAF therapy, which could provide rapid clinical benefit. Approximately 80% of patients had PD-L1-positive tissue samples. - At the time of the second interim analysis, the OS results crossed the prespecified efficacy boundary: the two pembrolizumab groups were superior to the OS in the ipilimumab group at the prespecified, one-sided, alpha level of 0.005. The Data and Monitoring Safety Board (DMSB) recommended that the study results be unblinded and pembrolizumab be made available to patients with disease progression in the ipilimumab group. The median duration of follow-up at time of second interim analysis was 12 months for all patients, with 289 deaths. Median OS had not been reached in any treatment group.
- The 1-year estimates of survival were 74% for patients receiving pembrolizumab every 2 weeks (hazard ratio [HR]death as compared with the ipilimumab group, 0.63; 95% CI, 0.47-0.83; P < .0005); 68% for patients receiving pembrolizumab every 3 weeks (HRdeath compared with ipilimumab, 0.69; 95% CI, 0.52-0.90; P = .0036); and 58% for patients treated with ipilimumab.
- Benefit was seen across all subgroups with the exception of patients with PD-L1-negative tumors. However, since this subset was small (18% of patients) and the CI was wide, no definitive conclusions could be drawn from this study.
- Grade 3 to 5 AEs considered to be related to a study drug were 13% for pembrolizumab every 2 weeks, 10% for pembrolizumab every 3 weeks, and 20% for ipilimumab. Rates of study-drug discontinuation as a result of toxicity were 4% for pembrolizumab every 2 weeks, 7% for pembrolizumab every 3 weeks, and 9% for ipilimumab. The one death, resulting from cardiac arrest, occurred in the ipilimumab group in a patient with type 2 diabetes mellitus caused by metabolic imbalances associated with ipilimumab-induced diarrhea.
Nivolumab Evidence (nivolumab): - Previously treated patients. Accelerated approval was based on a planned noncomparative interim analysis of the first 120 patients who received nivolumab with at least 6 months' follow-up from a multicenter, open-label trial (CheckMate 037 [NCT01721746]) that randomly assigned patients (2:1) to nivolumab (3mg/kg every 2 weeks) or the investigator's choice of chemotherapy (either DTIC 1,000 mg/m2 IV every 3 weeks or the combination of carboplatin [area under the curve 6] plus paclitaxel 175 mg/m2 every 3 weeks).[FDA label][Level of evidence: 3iiiDiv] Patients were required to have unresectable or metastatic melanoma that had progressed after treatment with ipilimumab and, if BRAF V600 mutation-positive, a BRAF inhibitor. The trial excluded patients with an autoimmune disease, a condition requiring immunosuppression, or a history of severe irAEs from treatment with ipilimumab.
- Median age of patients was 58 years; 65% of patients were male; and, ECOG PS was 0 in 58% of patients. BRAF V600 mutation was present in 22% of patients; 76% had M1c disease; 56% had an elevated LDH; 18% had a history of brain metastases; and, 68% had received two or more systemic therapies previously for metastatic disease.
- ORR and OS were coprimary endpoints. ORR was 32% (95% CI, 23-41) with four CRs and 34 PRs as assessed by RECIST 1.1 criteria and an independent central review. Among the 38 patients with responses, 33 (87%) had ongoing responses with durations from 2.6+ to 10.0+ months.
- Responses were seen in patients with and without BRAF V600 mutations.
- Safety analysis is based on 268 patients. Nivolumab was discontinued because of AEs in 9% of patients. Serious AEs occurred in 41% of patients and grade 3 and grade 4 AEs occurred in 42% of patients. The most common AEs were rash, cough, upper respiratory tract infection, and peripheral edema. Other important AEs included ventricular arrhythmia, iridocyclitis, increased amylase and lipase, dizziness, and neuropathy.
The FDA label provides recommendations for suspected irAEs, including withholding the drug and administering corticosteroids. - Previously untreated patients. A total of 418 patients with unresectable stage III or stage IV melanoma without a BRAF mutation were randomly assigned (1:1) in a double-blind multicenter trial to receive nivolumab (3 mg/kg every 2 weeks) and a DTIC-matched placebo (every 3 weeks) or DTIC (1,000 mg/m2 every 3 weeks with a nivolumab-matched placebo every 2 weeks). The primary endpoint was OS.[4][Level of evidence: 1iA ] The trial was conducted in 80 centers in Europe, Israel, Australia, Canada, and South America, which are countries where DTIC had been a standard first-line treatment in patients without a BRAF mutation.
- The DMSB noted a potential difference in OS during safety review. On June 10, 2014, an abbreviated report from an unplanned interim-database lock was reviewed showing a significant difference in OS in favor of nivolumab. The DMSB recommended that the study be unblinded and allow patients on DTIC to receive nivolumab. The intended sample size was approximately 410 patients; a total of 418 patients had been entered.
- Results from the double-blind portion of the study before the crossover amendment showed that median OS was not reached in the nivolumab group and was 10.8 months (95% CI, 9.3-12.1) in the DTIC group. The OS rate at 1 year was 72.9% (95% CI, 65.5-78.9) in the nivolumab group and 42.1% (95% CI, 33.0-50.9) in the dacarbazine group. The HRdeath was 0.42; 99.79% CI, 0.25-0.73; P < .001.
- The most common AEs in the nivolumab group were fatigue (19.9%), pruritus (17%), nausea (16.5%), and diarrhea (16%). In the nivolumab group, 6.8% of patients discontinued study treatment because of AEs compared with 11.7% of patients discontinuing study treatment in the DTIC group. AEs with potential immunologic etiology that occurred included gastrointestinal, hepatic, pulmonary, renal, endocrine, and skin; however, the majority resolved with a delay in study treatment, glucocorticoid administration, or both per management guidelines for nivolumab. No deaths were attributed to drug-related AEs in either group.
Anti-CTLA-4 Ipilimumab Ipilimumab is a human monoclonal antibody that binds to CTLA-4, thereby blocking its ability to down-regulate T-cell activation, proliferation, and effector function. Ipilimumab has demonstrated clinical benefit by prolonging OS in randomized trials, and was approved by the FDA in 2011. Two prospective, randomized, international trials, one each in previously untreated and treated patients, supported the use of ipilimumab.[5,6] Evidence (ipilimumab): - Previously treated patients: A total of 676 patients with previously treated, unresectable stage III or stage IV disease, and who were HLA-A*0201-positive, were entered into a three-arm, multinational, randomized (3:1:1), double-blind, double-dummy trial. A total of 403 patients were randomly assigned to receive ipilimumab (3 mg/kg every 3 weeks for 4 doses) with glycoprotein 100 (gp100) peptide vaccine. One hundred thirty-seven patients received ipilimumab (3 mg/kg every 3 weeks for 4 doses), and 136 patients received the gp100 vaccine. Patients were stratified by baseline metastases and previous receipt or nonreceipt of IL-2 therapy. Eighty-two of the patients had metastases to the brain at baseline.[6][Level of evidence: 1iA]
- The median OS was 10 months among patients who received ipilimumab alone and 10.1 months among those receiving ipilimumab with the gp100 vaccine, compared with 6.4 months for patients who received the vaccine alone (HR of ipilimumab alone vs. gp100 alone, 0.66; P < .003; HR of ipilimumab plus vaccine vs. gp100 alone, 0.68; P < .001).
- An analysis at 1 year showed that among patients treated with ipilimumab, 44% of those treated with ipilimumab and 45% of those treated with ipilimumab and the vaccine were alive, compared with 25% of the patients who received the vaccine only.
- Grade 3 or grade 4 irAEs occurred in 10% to 15% of patients treated with ipilimumab. These irAEs most often included diarrhea or colitis, and endocrine-related events (e.g., inflammation of the pituitary). These events required cessation of therapy and institution of anti-inflammatory agents such as corticosteroids or, in four cases, infliximab (an antitumor necrosis factor-alpha antibody).
- There were 14 drug-related deaths (2.1%), and seven deaths were associated with irAEs.
- Previously untreated patients: A multicenter, international trial randomly assigned 502 patients untreated for metastatic disease (adjuvant treatment was allowed) in a 1:1 ratio to receive ipilimumab (10 mg/kg) plus DTIC (850 mg/m2) or placebo plus DTIC (850 mg/m2) at weeks 1, 4, 7, and 10 followed by DTIC alone every 3 weeks through week 22. Patients with stable disease or an objective response and no dose-limiting toxic effects received ipilimumab or placebo every 12 weeks thereafter as maintenance therapy. The primary endpoint was survival. Patients were stratified according to ECOG PS and metastatic stage. Approximately 70% of the patients had an ECOG PS of 0, and the remainder of the patients had an ECOG PS of 1. Approximately 55% of patients had stage M1c disease.[5][Level of evidence: 1iA]
- The median OS was 11.2 months (95% CI, 9.4-13.6) for the ipilimumab-DTIC group versus 9.1 months (95% CI, 7.8-10.5) for the placebo-DTIC group. Estimated survival rates in the ipilimumab-DTIC group were 47.3% at 1 year, 28.5% at 2 years, and 20.8% at 3 years (HRdeath, 0.72; P < .001); and in the placebo-DTIC group, the rates were 36.3% at 1 year, 17.9% at 2 years, and 12.2% at 3 years.
- The most common study-drug-related AEs were those classified as immune related. Grade 3 or grade 4 irAEs were seen in 38.1% of patients treated with ipilimumab plus DTIC versus 4.4% of patients treated with placebo plus DTIC, the most common events were hepatitis and enterocolitis.
- No drug-related deaths occurred.
Clinicians and patients should be aware that immune-mediated adverse reactions may be severe or fatal. Early identification and treatment, including potential administration of systemic glucocorticoids or other immunosuppressants according to the immune-mediated adverse reaction management guide provided by the manufacturer, are necessary.[7] High-dose IL-2 IL-2 was approved by the FDA in 1998 on the basis of durable CRs in eight phase I and II studies. Phase III trials comparing high-dose IL-2 to other retreatments, providing an assessment of relative impact on OS, have not been conducted. Evidence (high-dose IL-2): - Based on a pooled analysis of 270 patients from eight single- and multi-institutional trials in 22 institutions conducted between 1985 and 1993:
- High-dose IL-2 demonstrated a 6% to 7% CR rate.[8]
- With a median follow-up time for surviving patients of at least 7 years, the median duration of CRs was not reached but was at least 59 months.[9]
Strategies to improve this therapy are an active area of investigation. Dual immunomodulation T-cells coexpress several receptors that inhibit T-cell function. Preclinical data and early clinical data suggest that coblockade of the two inhibitory receptors, CTLA-4 and PD-1, may be more effective than blockade of either alone.[10] This has led to a phase III trial (NCT01844505) comparing each single agent with the combination. Dual checkpoint inhibition CTLA-4 inhibitor plus PD-1 inhibitor Evidence (ipilimumab plus nivolumab): - Previously untreated patients. In an international, randomized, double-blind trial, 945 previously untreated patients with unresectable stage III or IV melanoma were randomly assigned in a 1:1:1 ratio to:
- ARM 1: nivolumab alone 3 mg/kg q 2 weeks plus placebo;
- ARM 2: nivolumab (1 mg/kg q 3 weeks) plus ipilimumab (3 mg/kg q 3 weeks for 4 doses) followed by 3 mg of nivolumab q 2 weeks; or
- ARM 3: ipilimumab alone (3 mg/kg q 3 weeks for 4 doses plus placebo).
PFS and OS were coprimary endpoints. Patients were stratified according to tumor PD-L1 status assessed in a central laboratory by immunohistochemical testing (positive vs. negative or indeterminate), BRAF mutation status (V600 mutation-positive vs. wild-type) and American Joint Committee on Cancer stage.[11][Level of evidence: 1iiA]
- Characteristics at baseline included 74% of patients with an ECOG PS of 0; 36% had elevated LDH; 31.5% had a BRAF mutation; and 58% had M1c disease. A minority of patients (23.6%) had a positive tumor PD-L1.
- Treatment with nivolumab alone or in combination with ipilimumab resulted in significantly longer PFS than with ipilimumab alone. Results were consistent across the prespecified stratification factors. Median PFS was 6.9 months (95% CI, 4.3-9.5) with nivolumab, 11.5 months (95% CI, 8.9-16.7) with nivolumab plus ipilimumab, and 2.9 months (95% CI, 2.8-3.4) with ipilimumab. The prespecified statistical analysis provided for a two-sided log-rank comparison of the combination and nivolumab monotherapy to ipilimumab, but not for a comparison of nivolumab with the combination. The HRdeath or disease progression for the combination versus ipilimumab was 0.42 (99.5% CI, 0.31-0.57; P < .001); for nivolumab versus ipilimumab, the HR was 0.57; 99.5% CI, 0.43-0.76; P < .001).
- AEs were highest in the combination arm and need to be monitored carefully. Grade 3 to 4 treatment-related AEs occurred in 16.3% of patients in the nivolumab group, 27.3% of patients in the ipilimumab group, and 55% of patients in the combination group. The most frequent reason for treatment discontinuation was disease progression in the two monotherapy arms-49% with nivolumab and 65% with ipilimumab. The most frequent reason for discontinuation in the combination group was toxicity (38%).
- The use of tumor PD-L1 status as a biomarker is an area of active research. OS from this study is not yet available, and the most effective assay and cutoff point remain to be determined.
Signal-transduction inhibitors Studies to date indicate that both BRAF and MEK (mitogen-activated ERK-[extracellular signal-regulated kinase] activating kinase) inhibitors, as single agents and in combination, can significantly impact the natural history of melanoma, although they do not appear to provide a cure. BRAFinhibitors Vemurafenib Vemurafenib is an orally available, small molecule, selective BRAF kinase inhibitor that was approved by the FDA in 2011 for patients with unresectable or metastatic melanoma who test positive for the BRAF V600E mutation. Treatment with vemurafenib is discouraged in wild-type BRAF melanoma because data from preclinical models have demonstrated that BRAF inhibitors can enhance rather than down-regulate the MAPK pathway in tumor cells with wild-type BRAF and upstream RAS mutations.[12,13,14,15] Evidence (vemurafenib): - Previously untreated patients: The approval of vemurafenib was supported by an international, multicenter trial (BRIM-3 [NCT01006980]) that screened 2,107 patients with previously untreated stage IIIC or IV melanoma for the BRAF V600 mutation and identified 675 patients via the cobas 4800 BRAF V600 Mutation Test.[16] Patients were randomly assigned to receive either vemurafenib (960 mg orally twice daily) or DTIC (1,000 mg/m2 IV every 3 weeks). Coprimary endpoints were rates of OS and PFS. At the planned interim analysis, the DMSB determined that both the OS and PFS endpoints had met the prespecified criteria for statistical significance in favor of vemurafenib and recommended that patients in the DTIC group be allowed to cross over to receive vemurafenib.[16][Levels of evidence: 1iiA and 1iiDiii]
- A total of 675 patients were evaluated for OS; although the median survival had not yet been reached for vemurafenib and the data were immature for reliable Kaplan-Meier estimates of survival curves, the OS in the vemurafenib arm was clearly superior to that in the DTIC arm.
- The HRdeath in the vemurafenib group was 0.37 (95% CI, 0.26-0.55; P < .001). The survival benefit in the vemurafenib group was observed in each prespecified subgroup, for example, age, sex, ECOG PS, tumor stage, LDH, and geographic region.
- The HR for tumor progression in the vemurafenib arm was 0.26 (95% CI, 0.20-0.33; P < .001). The estimated median PFS was 5.3 months in the vemurafenib arm versus 1.6 months in the DTIC arm.
- Twenty patients had non-BRAF V600E mutations: 19 with BRAF V600K and 1 with BRAF V600D. Four patients with a BRAF V600K mutation had a response to vemurafenib.
- AEs required dose modification or interruption in 38% of patients receiving vemurafenib and 16% of those receiving DTIC. The most common AEs with vemurafenib were cutaneous events (i.e., arthralgia and fatigue). Cutaneous squamous cell carcinoma (SCC), keratoacanthoma, or both developed in 18% of patients and were treated by simple excision. The most common AEs with DTIC were fatigue, nausea, vomiting, and neutropenia. (Refer to the PDQ summaries on Supportive and Palliative Care for more information on coping with cancer.)
- Previously treated patients: A total of 132 patients with a BRAF V600E or BRAF V600K mutation were enrolled in a multicenter phase II trial of vemurafenib, which was administered as 960 mg orally twice daily. Of the enrolled patients, 61% had stage M1c disease, and 49% had an elevated LDH level. All patients had received one or more previous therapies for advanced disease. Median follow-up was 12.9 months.[17][Level of evidence: 3iiiDiv]
- An independent review committee (IRC) reported a 53% response rate (95% CI, 44-62), with eight patients (6%) achieving CR.
- Median duration of response per IRC assessment was 6.7 months (95% CI, 5.6-8.6). Most responses were evident at the first radiologic assessment at 6 weeks; however, some patients did not respond until after receiving therapy for more than 6 months.
Dabrafenib Dabrafenib is an orally available, small molecule, selective BRAF inhibitor that was approved by the FDA in 2013 for patients with unresectable or metastatic melanoma who test positive for the BRAF V600E mutation as detected by an FDA-approved test. Dabrafenib and other BRAF inhibitors are not recommended for treatment of BRAF wild-type melanomas, as in vitro experiments suggest there may be a paradoxical stimulation of MAPK signaling resulting in tumor promotion. Evidence (dabrafenib): - An international, multicenter trial (BREAK-3 [NCT01227889]) compared dabrafenib with DTIC. A total of 250 patients with unresectable stage III or IV melanoma and BRAF V600E mutations were randomly assigned in a 3:1 ratio (dabrafenib 150 mg orally twice a day or DTIC 1,000 mg/m2 IV every 3 weeks). IL-2 was allowed as a previous treatment for advanced disease. The primary endpoint was PFS; patients could cross over at the time of progressive disease after confirmation by a blinded IRC.[18][Level of Evidence: 1iiDiii]
- With 126 events, the HR for PFS was 0.30 (95% CI, 0.18-0.51; P < .0001). The estimated median PFS was 5.1 months for dabrafenib versus 2.7 months for DTIC. OS data are limited by the median duration of follow-up and crossover. The PR rate was 47% versus 5%, and CR was 3% versus 2% in patients receiving dabrafenib versus DTIC, respectively.
- The most frequent AEs in patients treated with dabrafenib were cutaneous findings (i.e., hyperkeratosis, papillomas, palmar-plantar erythrodysesthesia), pyrexia, fatigue, headache, and arthralgia. Cutaneous SCC or keratoacanthoma occurred in 12 patients, basal cell carcinoma occurred in four patients, mycosis fungoides occurred in one patient, and new melanoma occurred in two patients.
MEKinhibitors Trametinib Trametinib is an orally available, small-molecule, selective inhibitor of MEK1 and MEK2. BRAF activates MEK1 and MEK2 proteins, which in turn, activate MAPK. Preclinical data suggest that MEK inhibitors can restrain growth and induce cell death of some BRAF-mutated human melanoma tumors. BRAF activates MEK1 and MEK2 proteins, which, in turn, activate MAPK. In 2013, trametinib was approved by the FDA for patients with unresectable or metastatic melanoma with BRAF V600E or K mutations, as determined by an FDA-approved test. Evidence (trametinib): - A total of 1,022 patients were screened for BRAF mutations, resulting in 322 eligible patients (281 with BRAF V600E, 40 with BRAF V600K, and one with both mutations).[19] One previous treatment (biologic or chemotherapy) was allowed; however, no previous treatment with a BRAF or MEK inhibitor was permitted. Patients were randomly assigned in a 2:1 ratio to receive trametinib (2 mg once daily) or IV chemotherapy (either DTIC 1,000 mg/m2 every 3 weeks or paclitaxel 175 mg/m2 every 3 weeks). Crossover for patients randomly assigned to chemotherapy was allowed; therefore, the primary endpoint was PFS.
- The investigator-assessed PFS was 4.8 months in patients receiving trametinib versus 1.5 months in the chemotherapy group (HR for PFS or death, 0.45; 95% CI, 0.33-0.63; P < .001). A radiology review blinded-to-treatment arm resulted in similar outcomes. Median OS has not been reached.
- AEs leading to dose interruptions occurred in 35% of patients in the trametinib group and 22% of those in the chemotherapy group. AEs leading to dose reductions occurred in 27% of patients who received trametinib and in 10% of those who received chemotherapy.
- The most common AEs included rash, diarrhea, nausea, vomiting, fatigue, peripheral edema, alopecia, hypertension, and constipation. Cardiomyopathy (7%), interstitial lung disease (2.4%), central serous retinopathy (<1%), and retinal-vein occlusion (<1%) are uncommon but serious AEs associated with trametinib. On-study cutaneous SCCs were not observed. (Refer to the PDQ summaries on Supportive and Palliative Care for more information on coping with cancer.)
Cobimetinib Cobimetinib is a small-molecule, selective MEK inhibitor that was approved by the FDA in 2015 for use in combination with the BRAF inhibitor vemurafenib. (Refer to the Combination therapy with signal-transduction inhibitors section of this summary for more information.) KITinhibitors Early data suggest that mucosal or acral melanomas with activating mutations or amplifications in c-KIT may be sensitive to a variety of c-KIT inhibitors.[20,21,22] Phase II and phase III trials are available for patients with unresectable stage III or stage IV melanoma harboring the c-KIT mutation. Multikinase inhibitors Sorafenib The multikinase inhibitor sorafenib has activity against both the vascular endothelial growth-factor signaling and the Raf/MEK/ERK pathway. This agent had minimal activity as a single agent in melanoma treatment. Two large, multicenter, placebo-controlled, randomized trials of carboplatin and paclitaxel plus or minus sorafenib showed no improvement over chemotherapy alone as either first-line treatment or second-line treatment.[18,23] Combination therapy with signal-transduction inhibitors Resistance to BRAF inhibitors, in patients with BRAF V600 mutations, may be associated with reactivation of the MAPK pathway. Combinations of signal-transduction inhibitors that block different sites in the same pathway or sites in multiple pathways are an active area of research. BRAF inhibitor plusMEKinhibitor Evidence (dabrafenib plus trametinib): - In January 2014, the FDA granted accelerated approval to dabrafenib and trametinib in combination to treat patients with unresectable or metastatic melanomas who carry the BRAF V600E or V600K mutation as detected by an FDA-approved test. Accelerated approval was granted on the basis of objective response rates from an open-label phase II trial that randomly assigned 162 patients with unresectable or metastatic melanoma with the BRAF V600E or V600K mutation in a 1:1:1 ratio to receive dabrafenib alone (150 mg twice a day) or with trametinib (at a dose of either 1 mg or 2 mg twice a day).[24] Patients who had disease progression on dabrafenib monotherapy could cross over to receive the combination of dabrafenib 150 mg plus trametinib 2 mg twice a day. Patients were allowed to have received one previous therapy other than a BRAF or MEK inhibitor.[24][Level of evidence: 1iiDiv].
- Patients treated with the combination had a response rate of 76%, with an average duration of 10.5 months. Patients treated with dabrafenib alone had a response rate of 54%, with an average duration of 5.6 months.
Full approval for the combination will depend on demonstration of improvements in PFS and survival from randomized trials. - Previously untreated. An international, double-blind, phase III trial without crossover randomly assigned 423 previously untreated patients with unresectable stage IIIC or stage IV melanoma with BRAF V600E or V600K mutations to receive the combination of dabrafenib (150 mg orally twice daily) plus trametinib (2 mg orally once daily) or dabrafenib plus placebo. The primary endpoint was investigator-assessed PFS. The protocol included a prespecified interim analysis for OS at the time of analysis of the primary endpoint. Patients were stratified by baseline LDH and BRAF genotype.[25]
- Median PFS was 9.3 months for the combination versus 8.8 months for dabrafenib plus placebo. The HRdeath or progression was 0.75 (95% CI, 0.57-0.99; P = .03). Updated data at the time of final analysis of OS revealed a median PFS of 11.0 months for the combination versus 8.8 months for dabrafenib plus placebo. The HR for PFS or death was 0.67 (95% CI, 0.53-0.84; P = .0004; unadjusted for multiple testing).[26]
- A prespecified final analysis of OS was conducted at 70% of events. Median OS was 25.1 months in the dabrafenib-plus-trametinib group (66% events) versus 18.7 months in the dabrafenib-plus-placebo group (76% events). The HR was 0.71 (95% CI, 0.55-0.92; P = 0.01).
- Permanent discontinuations of study drugs were reported in 9% of patients on the combination and in 5% of patients treated with dabrafenib only.
- The incidence of grade 3 to grade 4 AEs was similar between the groups: a 35% incidence with the combination and a 37% incidence with dabrafenib only. Pyrexia occurred more frequently with the combination and was treated with immediate temporary cessation of the study drug in either group; prophylactic glucocorticoids may prevent recurring episodes. Hyperproliferative cutaneous events, including cutaneous SCC, which is considered related to paradoxical activation of the MAPK pathway, occurred less frequently with the addition of the MEK inhibitor. Rare, but serious, AEs included decreased ejection fraction and chorioretinopathy.
- Previously untreated. An international, open-label, phase III trial randomly assigned 704 previously untreated patients with metastatic melanoma with a BRAF V600 mutation to receive standard doses of either the combination of dabrafenib plus trametinib or vemurafenib as first-line therapy. The primary endpoint was OS.[27][Level of evidence: 1iiA]
- An interim analysis for OS was planned when 202 of the final 288 events occurred. Per protocol, the DMSB used adjusted efficacy boundaries for actual events (222) (2-sided P < .0214 for efficacy and P > .2210 for futility). The DMSB recommended stopping for efficacy, and the interim analysis is considered to be the final analysis of OS. A protocol amendment was issued to allow crossover to the combination therapy arm.
- A total of 100 patients (28%) in the combination arm and 122 (35%) in the vemurafenib group had died (HR, 0.69; 95% CI, 0.53-0.89; P = .005). Median OS for patients treated with vemurafenib was 17.2 months; the median has not been reached in the combination therapy arm.
Evidence (vemurafenib plus cobimetinib): - An international phase III trial randomly assigned 495 patients with previously untreated, unresectable stage IIIC or stage IV melanoma with BRAF V600 mutation-positive melanoma to receive the combination of vemurafenib (960 mg orally twice daily) and cobimetinib (60 mg orally once daily for 21 days followed by a 7-day rest period) or vemurafenib plus placebo. The primary endpoint was investigator-assessed PFS. Crossover at time of PFS was not allowed. Patients were stratified by stage and geographic region. Two interim analyses of OS were prespecified, with the first specified at the time of analysis of the primary endpoint.[28][Level of evidence: 1iDiii]
- Median PFS was 9.9 months for the combination versus 6.2 months in patients treated with vemurafenib plus placebo. The HRdeath or progression was 0.51 (95% CI, 0.39-0.68; P = .001).
- The first interim analysis of OS is immature because of the few events in both arms; therefore, median survival was not reached in either study group.
- Rate of withdrawal of therapy caused by AEs was similar between the groups: a 13% rate was found with patients treated with the combination, and a 12% rate was found with patients treated with vemurafenib only. Six deaths were attributed to AEs in the combination group, and three deaths were attributed to AEs in the vemurafenib only group.
- The incidence of grade 3 to grade 4 AEs was similar between the groups: a 62% incidence rate was found in patients treated with the combination, and a 58% rate was found in patients treated with vemurafenib alone. Rare, but serious, AEs included chorioretinopathy, retinal detachment, decreased ejection fraction, and QT prolongation. Hyperproliferative cutaneous events, including cutaneous SCC, which was considered to be related to paradoxical activation of the MAPK pathway, occurred less frequently with the addition of the MEK inhibitor.
Chemotherapy DTIC was approved in 1970 on the basis of ORRs. Phase III trials indicate an ORR of 10% to 20%, with rare CRs observed. An impact on OS has not been demonstrated in randomized trials.[5,16,29,30,31] When used as a control arm for recent registration trials of ipilimumab and vemurafenib in previously untreated patients with metastatic melanoma, DTIC was shown to be inferior for OS. Temozolomide, an oral alkylating agent that hydrolyzes to the same active moiety as DTIC, appeared to be similar to DTIC (IV administration) in a randomized, phase III trial with a primary endpoint of OS; however, the trial was designed for superiority, and the sample size was inadequate to prove equivalency.[30] The objective response rate to DTIC and the nitrosoureas, carmustine and lomustine, is approximately 10% to 20%.[29,32,33,34] Responses are usually short-lived, ranging from 3 to 6 months, although long-term remissions can occur in a limited number of patients who attain a CR.[32,34] A randomized trial compared IV DTIC with temozolomide, an oral agent; OS was 6.4 months for DTIC versus 7.7 months for temozolomide (HR, 1.18; 95% CI, 0.92-1.52). While these data suggested similarity between DTIC and temozolomide, no benefit in survival has been demonstrated for either DTIC or temozolomide; therefore, demonstration of similarity did not result in approval of temozolomide by the FDA.[30][Level of evidence: 1iiA] An extended schedule and escalated dose of temozolomide was compared with DTIC in a multicenter trial by the European Organisation for Research and Treatment of Cancer (EORTC)-18032 [NCT00101218] randomly assigning 859 patients. No improvement was seen in OS or PFS for the temozolomide group, and this dose and schedule resulted in more toxicity than standard-dose, single-agent DTIC.[35][Level of evidence: 1iiA] Two randomized, phase III trials in previously untreated patients with metastatic melanoma (resulting in FDA approval for vemurafenib [16] and ipilimumab [5]) included DTIC as the standard therapy arm. Both vemurafenib (in BRAF V600 mutant melanoma) and ipilimumab showed superior OS compared with DTIC in the two separate trials. Other agents with modest, single-agent activity include vinca alkaloids, platinum compounds, and taxanes.[32,33] Attempts to develop combination regimens that incorporate chemotherapy (e.g., multiagent chemotherapy,[36,37] combinations of chemotherapy and tamoxifen,[38,39,40] and combinations of chemotherapy and immunotherapy [8,9,36,41,42,43,44]) have not demonstrated an improvement in OS. A published data meta-analysis of 18 randomized trials (15 of which had survival information) that compared chemotherapy with biochemotherapy (i.e., the same chemotherapy plus interferon alone or with IL-2) reported no impact on OS.[45][Level of evidence:1iiA] Palliative local therapy Melanoma metastatic to distant, lymph node-bearing areas may be palliated by regional lymphadenectomy. Isolated metastases to the lung, gastrointestinal tract, bone, or sometimes the brain may be palliated by resection, with occasional long-term survival.[42,43,44] Although melanoma is a relatively radiation-resistant tumor, palliative radiation therapy may alleviate symptoms. Retrospective studies have shown that symptom relief and some shrinkage of the tumor with radiation therapy may occur in patients with the following:[46,47] - Multiple brain metastases.
- Bone metastases.
- Spinal cord compression.
The most effective dose-fractionation schedule for palliation of melanoma metastatic to the bone or spinal cord is unclear, but high-dose-per-fraction schedules are sometimes used to overcome tumor resistance. (Refer to the PDQ summary on Cancer Pain for more information.) Treatment Options Under Clinical Evaluation for Unresectable Stage III, Stage IV, and Recurrent Melanoma - Immunotherapy-single agent, and combination immunomodulation.
- Targeted therapy-single-agent and combination therapy.
- Signal-transduction inhibitors, including P13K (phosphoinositide-3 kinase) and Akt (protein kinase B) inhibitors, CDK (cyclin-dependent kinase) in addition to BRAF and MEK.
- Antiangiogenesis agents. Preclinical data suggest that increased vascular endothelial growth factor production may be implicated in resistance to BRAF inhibitors.[48]
- Targeted therapy for specific melanoma populations.
- In smaller subsets of melanoma, activating mutations may occur in NRAS (neuroblastoma RAS viral [v-ras] oncogene homolog) (15%-20%), c-KIT (28%-39% of melanomas arising in chronically sun-damaged skin, or acral and mucosal melanomas), and CDK4 (cyclin-dependent kinase 4) (<5%), whereas GNAQ is frequently mutated in uveal melanomas. Drugs developed to target the pathways activated by these mutations are currently in clinical trials.
- Combinations of immunotherapy and targeted therapy.
- Intralesional injections (for example, oncologic viruses).
- Complete surgical resection of all known disease versus best medical therapy.
- Isolated limb perfusion for unresectable extremity melanoma.
- Systemic therapy for unresectable disease.
Current Clinical Trials Check the list of NCI-supported cancer clinical trials that are now accepting patients with stage III melanoma, stage IV melanoma and recurrent melanoma. The list of clinical trials can be further narrowed by location, drug, intervention, and other criteria. General information about clinical trials is also available from the NCI website. References:
-
Andtbacka RH, Kaufman HL, Collichio F, et al.: Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 33 (25): 2780-8, 2015.
-
Robert C, Ribas A, Wolchok JD, et al.: Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384 (9948): 1109-17, 2014.
-
Robert C, Schachter J, Long GV, et al.: Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372 (26): 2521-32, 2015.
-
Robert C, Long GV, Brady B, et al.: Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372 (4): 320-30, 2015.
-
Robert C, Thomas L, Bondarenko I, et al.: Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364 (26): 2517-26, 2011.
-
Hodi FS, O'Day SJ, McDermott DF, et al.: Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711-23, 2010.
-
Yervoy (Ipilimumab): Serious and Fatal Immune-Mediated Adverse Reactions [Medication Guide]. Princeton, NJ: Bristol-Myers Squibb, 2011. Available online. Last accessed December 8, 2016.
-
Atkins MB, Lotze MT, Dutcher JP, et al.: High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17 (7): 2105-16, 1999.
-
Atkins MB, Kunkel L, Sznol M, et al.: High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am 6 (Suppl 1): S11-4, 2000.
-
Postow MA, Chesney J, Pavlick AC, et al.: Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372 (21): 2006-17, 2015.
-
Larkin J, Chiarion-Sileni V, Gonzalez R, et al.: Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373 (1): 23-34, 2015.
-
Heidorn SJ, Milagre C, Whittaker S, et al.: Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140 (2): 209-21, 2010.
-
Hatzivassiliou G, Song K, Yen I, et al.: RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464 (7287): 431-5, 2010.
-
Poulikakos PI, Zhang C, Bollag G, et al.: RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464 (7287): 427-30, 2010.
-
Su F, Viros A, Milagre C, et al.: RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366 (3): 207-15, 2012.
-
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011.
-
Sosman JA, Kim KB, Schuchter L, et al.: Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366 (8): 707-14, 2012.
-
Hauschild A, Grob JJ, Demidov LV, et al.: Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380 (9839): 358-65, 2012.
-
Flaherty KT, Robert C, Hersey P, et al.: Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367 (2): 107-14, 2012.
-
Hodi FS, Friedlander P, Corless CL, et al.: Major response to imatinib mesylate in KIT-mutated melanoma. J Clin Oncol 26 (12): 2046-51, 2008.
-
Guo J, Si L, Kong Y, et al.: Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29 (21): 2904-9, 2011.
-
Carvajal RD, Antonescu CR, Wolchok JD, et al.: KIT as a therapeutic target in metastatic melanoma. JAMA 305 (22): 2327-34, 2011.
-
Flaherty KT, Lee SJ, Zhao F, et al.: Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol 31 (3): 373-9, 2013.
-
Flaherty KT, Infante JR, Daud A, et al.: Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367 (18): 1694-703, 2012.
-
Long GV, Stroyakovskiy D, Gogas H, et al.: Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371 (20): 1877-88, 2014.
-
Long GV, Stroyakovskiy D, Gogas H, et al.: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386 (9992): 444-51, 2015.
-
Robert C, Karaszewska B, Schachter J, et al.: Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372 (1): 30-9, 2015.
-
Larkin J, Ascierto PA, Dréno B, et al.: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371 (20): 1867-76, 2014.
-
Chapman PB, Einhorn LH, Meyers ML, et al.: Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol 17 (9): 2745-51, 1999.
-
Middleton MR, Grob JJ, Aaronson N, et al.: Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18 (1): 158-66, 2000.
-
Avril MF, Aamdal S, Grob JJ, et al.: Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol 22 (6): 1118-25, 2004.
-
Anderson CM, Buzaid AC, Legha SS: Systemic treatments for advanced cutaneous melanoma. Oncology (Huntingt) 9 (11): 1149-58; discussion 1163-4, 1167-8, 1995.
-
Wagner JD, Gordon MS, Chuang TY, et al.: Current therapy of cutaneous melanoma. Plast Reconstr Surg 105 (5): 1774-99; quiz 1800-1, 2000.
-
Mays SR, Nelson BR: Current therapy of cutaneous melanoma. Cutis 63 (5): 293-8, 1999.
-
Patel PM, Suciu S, Mortier L, et al.: Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer 47 (10): 1476-83, 2011.
-
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996.
-
Kirkwood JM, Ibrahim JG, Sondak VK, et al.: High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18 (12): 2444-58, 2000.
-
Kirkwood JM, Ibrahim J, Lawson DH, et al.: High-dose interferon alfa-2b does not diminish antibody response to GM2 vaccination in patients with resected melanoma: results of the Multicenter Eastern Cooperative Oncology Group Phase II Trial E2696. J Clin Oncol 19 (5): 1430-6, 2001.
-
Hancock BW, Wheatley K, Harris S, et al.: Adjuvant interferon in high-risk melanoma: the AIM HIGH Study--United Kingdom Coordinating Committee on Cancer Research randomized study of adjuvant low-dose extended-duration interferon Alfa-2a in high-risk resected malignant melanoma. J Clin Oncol 22 (1): 53-61, 2004.
-
Koops HS, Vaglini M, Suciu S, et al.: Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. European Organization for Research and Treatment of Cancer Malignant Melanoma Cooperative Group Protocol 18832, the World Health Organization Melanoma Program Trial 15, and the North American Perfusion Group Southwest Oncology Group-8593. J Clin Oncol 16 (9): 2906-12, 1998.
-
Lee ML, Tomsu K, Von Eschen KB: Duration of survival for disseminated malignant melanoma: results of a meta-analysis. Melanoma Res 10 (1): 81-92, 2000.
-
Leo F, Cagini L, Rocmans P, et al.: Lung metastases from melanoma: when is surgical treatment warranted? Br J Cancer 83 (5): 569-72, 2000.
-
Ollila DW, Hsueh EC, Stern SL, et al.: Metastasectomy for recurrent stage IV melanoma. J Surg Oncol 71 (4): 209-13, 1999.
-
Gutman H, Hess KR, Kokotsakis JA, et al.: Surgery for abdominal metastases of cutaneous melanoma. World J Surg 25 (6): 750-8, 2001.
-
Ives NJ, Stowe RL, Lorigan P, et al.: Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. J Clin Oncol 25 (34): 5426-34, 2007.
-
Rate WR, Solin LJ, Turrisi AT: Palliative radiotherapy for metastatic malignant melanoma: brain metastases, bone metastases, and spinal cord compression. Int J Radiat Oncol Biol Phys 15 (4): 859-64, 1988.
-
Herbert SH, Solin LJ, Rate WR, et al.: The effect of palliative radiation therapy on epidural compression due to metastatic malignant melanoma. Cancer 67 (10): 2472-6, 1991.
-
Martin MJ, Hayward R, Viros A, et al.: Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov 2 (4): 344-55, 2012.
Changes to This Summary (01 / 26 / 2017)The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above. General Information About Melanoma Updated statistics with estimated new cases and deaths for 2017 (cited American Cancer Society as reference 1). This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages. About This PDQ SummaryPurpose of This Summary This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of melanoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions. Reviewers and Updates This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH). Board members review recently published articles each month to determine whether an article should: - be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary. The lead reviewers for Melanoma Treatment are: - Russell S. Berman, MD (New York University School of Medicine)
- Scharukh Jalisi, MD, FACS (Boston University Medical Center)
- Alison Martin, MD (Martin and Associates Consulting)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries. Levels of Evidence Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations. Permission to Use This Summary PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]." The preferred citation for this PDQ summary is: PDQ® Adult Treatment Editorial Board. PDQ Melanoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/skin/hp/melanoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389469] Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images. Disclaimer Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page. Contact Us More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us. Last Revised: 2017-01-26 Last modified on: 8 September 2017
|
|