Unusual Cancers of Childhood Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Unusual Cancers of Childhood Treatment (PDQ®): Treatment - Health Professional Information [NCI]Skip to the navigationGeneral Information About Unusual Cancers of ChildhoodIntroduction Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[1] Referral to medical centers with multidisciplinary teams of cancer specialists experienced in treating cancers that occur in childhood and adolescence should be considered for children and adolescents with cancer. This multidisciplinary team approach incorporates the skills of the primary care physician, pediatric surgeons, radiation oncologists, pediatric medical oncologists/hematologists, rehabilitation specialists, pediatric nurse specialists, social workers, and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life. (Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.) Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[2] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients/families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapy for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website. Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[3] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.) Childhood cancer is a rare disease with about 15,000 cases diagnosed annually in the United States in individuals younger than 20 years.[4] The U.S. Rare Diseases Act of 2002 defines a rare disease as one that affects populations smaller than 200,000 persons and, by definition, all pediatric cancers are considered rare. The designation of a pediatric rare tumor is not uniform among international groups: - The Italian cooperative project on rare pediatric tumors (Tumori Rari in Eta Pediatrica [TREP]) defines a pediatric rare tumor as one with an incidence of less than two cases per 1 million population per year and is not included in other clinical trials.[5]
- The Children's Oncology Group (COG) has opted to define rare pediatric cancers as those listed in the International Classification of Childhood Cancer subgroup XI, which includes thyroid cancer, melanoma and nonmelanoma skin cancers, and multiple types of carcinomas (e.g., adrenocortical carcinoma, nasopharyngeal carcinoma, and most adult-type carcinomas such as breast cancer, colorectal cancer, etc.).[6] These diagnoses account for about 4% of cancers diagnosed in children aged 0 to 14 years, compared with about 20% of cancers diagnosed in adolescents aged 15 to 19 years (refer to Figures 1 and 2).[7] Most cancers within subgroup XI are either melanomas or thyroid cancer, with the remaining subgroup XI cancer types accounting for only 1.3% of cancers in children aged 0 to 14 years and 5.3% of cancers in adolescents aged 15 to 19 years.
These rare cancers are extremely challenging to study because of the low incidence of patients with any individual diagnosis, the predominance of rare cancers in the adolescent population, and the lack of clinical trials for adolescents with rare cancers such as melanoma.
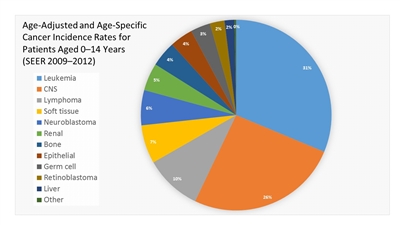
Figure 1. Age-adjusted and age-specific (0-14 years) Surveillance, Epidemiology, and End Results cancer incidence rates from 2009 to 2012 by International Classification of Childhood Cancer group and subgroup and age at diagnosis, including myelodysplastic syndrome and group III benign brain/central nervous system tumors for all races, males, and females.
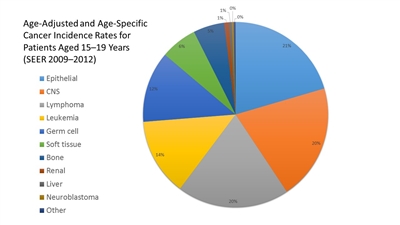
Figure 2. Age-adjusted and age-specific (15-19 years) Surveillance, Epidemiology, and End Results cancer incidence rates from 2009 to 2012 by International Classification of Childhood Cancer group and subgroup and age at diagnosis, including myelodysplastic syndrome and group III benign brain/central nervous system tumors for all races, males, and females.
Several initiatives to study rare pediatric cancers have been developed by the COG and other international groups, such as the International Society of Paediatric Oncology (Société Internationale D'Oncologie Pédiatrique [SIOP]). The Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) rare tumor project was founded in Germany in 2006.[8] The Italian cooperative project on rare pediatric tumors (TREP) was launched in 2000,[5] and the Polish Pediatric Rare Tumor Study Group was launched in 2002.[9] In Europe, the rare tumor studies groups from France, Germany, Italy, Poland, and the United Kingdom have joined in the European Cooperative study Group on Pediatric Rare Tumors (EXPeRT), focusing on international collaboration and analyses of specific rare tumor entities.[10] Within the COG, efforts have concentrated on increasing accrual to the COG registry (now known as the Childhood Cancer Research Network/Project Every Child) and the rare tumor bank, developing single-arm clinical trials, and increasing cooperation with adult cooperative group trials.[11] The accomplishments and challenges of this initiative have been described in detail.[6,12] The tumors discussed in this summary are very diverse; they are arranged in descending anatomic order, from infrequent tumors of the head and neck to rare tumors of the urogenital tract and skin. All of these cancers are rare enough that most pediatric hospitals might see less than a handful of some histologies in several years. The majority of the histologies described here occur more frequently in adults. Information about these tumors may also be found in sources relevant to adults with cancer. References:
-
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
-
Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004.
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
-
Ferrari A, Bisogno G, De Salvo GL, et al.: The challenge of very rare tumours in childhood: the Italian TREP project. Eur J Cancer 43 (4): 654-9, 2007.
-
Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children's Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010.
-
Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2012. Bethesda, Md: National Cancer Institute, 2015. Also available online. Last accessed February 15, 2017.
-
Brecht IB, Graf N, Schweinitz D, et al.: Networking for children and adolescents with very rare tumors: foundation of the GPOH Pediatric Rare Tumor Group. Klin Padiatr 221 (3): 181-5, 2009 May-Jun.
-
Balcerska A, Godziński J, Bień E, et al.: [Rare tumours--are they really rare in the Polish children population?]. Przegl Lek 61 (Suppl 2): 57-61, 2004.
-
Bisogno G, Ferrari A, Bien E, et al.: Rare cancers in children - The EXPeRT Initiative: a report from the European Cooperative Study Group on Pediatric Rare Tumors. Klin Padiatr 224 (6): 416-20, 2012.
-
Musselman JR, Spector LG, Krailo MD, et al.: The Children's Oncology Group Childhood Cancer Research Network (CCRN): case catchment in the United States. Cancer 120 (19): 3007-15, 2014.
-
Pappo AS, Furman WL, Schultz KA, et al.: Rare Tumors in Children: Progress Through Collaboration. J Clin Oncol 33 (27): 3047-54, 2015.
Head and Neck CancersChildhood sarcomas often occur in the head and neck area and they are described in other sections. Unusual pediatric head and neck cancers include the following: - Nasopharyngeal carcinoma.
- Esthesioneuroblastoma.
- Thyroid tumors.
- Oral cavity cancer.
- Salivary gland tumors.
- Laryngeal carcinoma and papillomatosis.
- Midline tract carcinoma involving the NUT gene.
The prognosis, diagnosis, classification, and treatment of these head and neck cancers are discussed below. It must be emphasized that these cancers are seen very infrequently in patients younger than 15 years, and most of the evidence is derived from small case series or cohorts combining pediatric and adult patients. Nasopharyngeal Carcinoma Incidence Nasopharyngeal carcinoma arises in the lining of the nasal cavity and pharynx, and it accounts for about one-third of all cancers of the upper airways in children.[1,2] Nasopharyngeal carcinoma is very uncommon in children younger than 10 years but increases in incidence to 0.8 cases per 1 million per year in children aged 10 to 14 years and 1.3 cases per million per year in children aged 15 to 19 years.[3,4,5] The incidence of nasopharyngeal carcinoma is characterized by racial and geographic variations, with an endemic distribution among well-defined ethnic groups, such as inhabitants of some areas in North Africa and the Mediterranean basin, and, particularly, Southeast Asia. In the United States, the incidence of nasopharyngeal carcinoma is higher in black children and adolescents younger than 20 years.[4] Risk Factors Nasopharyngeal carcinoma is strongly associated with Epstein-Barr virus (EBV) infection. In addition to the serological evidence of infection in more than 98% of patients, EBV DNA is present as a monoclonal episome in the nasopharyngeal carcinoma cells, and tumor cells can have EBV antigens on their cell surface.[6] The circulating levels of EBV DNA and serologic documentation of EBV infection may aid in the diagnosis.[7] Specific HLA subtypes, such as the HLA A2Bsin2 haplotype, are associated with a higher risk of nasopharyngeal carcinoma.[1] Histology Three histologic subtypes of nasopharyngeal carcinoma are recognized by the World Health Organization (WHO): - Type I is keratinizing squamous cell carcinoma.
- Type II is nonkeratinizing squamous cell carcinoma. Type II is distinguished into type IIa and IIb depending on the presence of lymphoid infiltration.
- Type III is undifferentiated carcinoma. Type III is distinguished into type IIIa and IIIb depending on the presence of lymphoid infiltration.
Children with nasopharyngeal carcinoma are more likely to have WHO type II or type III disease.[4] Clinical Presentation Signs and symptoms of nasopharyngeal carcinoma are as follows:[2,8] - Cervical lymphadenopathy.
- Nosebleeds.
- Nasal congestion and obstruction.
- Headache.
- Otalgia.
- Otitis media.
Given the rich lymphatic drainage of the nasopharynx, bilateral cervical lymphadenopathy is often the first sign of disease. The tumor spreads locally to adjacent areas of the oropharynx and may invade the skull base, resulting in cranial nerve palsy or difficulty with movements of the jaw (trismus). Distant metastatic sites may include the bones, lungs, and liver. Diagnostic and Staging Evaluation Diagnostic tests will determine the extent of the primary tumor and the presence of metastases. Visualization of the nasopharynx by an ear-nose-throat specialist using nasal endoscopy and magnetic resonance imaging of the head and neck can be used to determine the extent of the primary tumor. A diagnosis can be made from a biopsy of the primary tumor or enlarged lymph nodes of the neck. Nasopharyngeal carcinomas must be distinguished from all other cancers that can present with enlarged lymph nodes and from other types of cancer in the head and neck area. Thus, diseases such as thyroid cancer, rhabdomyosarcoma, non-Hodgkin lymphoma, Hodgkin lymphoma, and Burkitt lymphoma must be considered, as well as benign conditions such as nasal angiofibroma, which usually presents with epistaxis in adolescent males, infectious lymphadenitis, and Rosai-Dorfman disease. Evaluation of the chest and abdomen by computed tomography (CT) and bone scan is performed to determine whether there is metastatic disease. Fludeoxyglucose positron emission tomography (PET)-CT may also be helpful in the evaluation of potential metastatic lesions.[9] Staging Tumor staging is performed using the tumor-node-metastasis (TNM) classification system of the American Joint Committee on Cancer (AJCC, 7th edition).[10] More than 90% of children and adolescents with nasopharyngeal carcinoma present with advanced disease (stage III/IV or T3/T4).[11,12,13] Metastatic disease (stage IVC) at diagnosis is uncommon. A retrospective analysis of data from the Surveillance, Epidemiology, and End Results (SEER) program reported that patients younger than 20 years had a higher incidence of advanced-stage disease than did older patients.[4] Prognosis The overall survival (OS) of children and adolescents with nasopharyngeal carcinoma has improved over the last four decades; with state-of-the-art multimodal treatment, 5-year survival rates are in excess of 80%.[4,5,8,12,13,14,15] After controlling for stage, children with nasopharyngeal carcinoma have significantly better outcomes than do adults.[4,5] However, the intensive use of chemotherapy and radiation therapy results in significant acute and long-term morbidities, including subsequent neoplasms.[4,12,13,15] Treatment Treatment of nasopharyngeal carcinoma is multimodal and includes the following: - Combined-modality therapy with chemotherapy and radiation: High-dose radiation therapy alone has a role in the management of nasopharyngeal carcinoma; however, studies in both children and adults show that combined-modality therapy with chemotherapy and radiation is the most effective way to treat nasopharyngeal carcinoma.[12,13,14,16,17,18,19][Level of evidence: 2A]
- Randomized studies have investigated the role of chemotherapy in the treatment of adult nasopharyngeal carcinoma. The use of concomitant chemoradiation therapy was associated with a significant survival benefit, including improved locoregional disease control and reduction in distant metastases.[18,20] The use of neoadjuvant chemotherapy has also resulted in better local and distant control rates, whereas postradiation chemotherapy does not seem to offer any benefit.[20]
- In children, four studies used preradiation chemotherapy with different combinations of methotrexate, cisplatin, 5-fluorouracil (5-FU), and leucovorin with or without recombinant interferon-beta.[13,14,21,22][Level of evidence: 2A]
- These four studies reported response rates of more than 90% and excellent outcomes.
- Neoadjuvant chemotherapy with cisplatin and 5-FU (with or without leucovorin), followed by chemoradiation with single-agent cisplatin has yielded 5-year OS rates consistently above 80%.[13,14]
- A preliminary analysis of the NPC-2003-GPOH study, which included a 6-month maintenance therapy phase with interferon-beta, reported a 30-month OS estimate of 97.1%.[14]
- While nasopharyngeal carcinoma is a very chemosensitive neoplasm, high radiation doses to the nasopharynx and neck (approximately 60 Gy) are required for optimal locoregional control.[12,13,14,15] The combination of cisplatin-based chemotherapy and high doses of radiation therapy to the nasopharynx and neck are associated with a high probability of hearing loss, hypothyroidism and panhypopituitarism, trismus, xerostomia, dental problems, and chronic sinusitis or otitis.[12,13,15]; [8][Level of evidence: 3iiiA]
- A randomized prospective trial compared cisplatin and fluorouracil with cisplatin, fluorouracil, and docetaxel for the treatment of nasopharyngeal carcinoma in children and adolescents.[23][Level of evidence: 1iiA] The addition of docetaxel was not associated with improved outcome.
- Additional drug combinations that have been used in children with nasopharyngeal carcinoma include bleomycin with epirubicin and cisplatin, and cisplatin with methotrexate and bleomycin.[1]
- Other approaches to the management of nasopharyngeal carcinoma in children have been evaluated and include the following:
- Incorporation of high-dose-rate brachytherapy into the chemoradiation therapy approach.[24,25]
- Following adult studies and data, taxanes have been incorporated into the treatment of childhood nasopharyngeal carcinoma; studies have shown good objective response rates and favorable outcomes with the use of docetaxel in combination with cisplatin.[26][Level of evidence: 3iiiDiv]
- Surgery: Surgery has a limited role in the management of nasopharyngeal carcinoma because the disease is usually considered unresectable due to extensive local spread.
The use of EBV-specific cytotoxic T-lymphocytes has shown to be a very promising approach with minimal toxicity and evidence of significant antitumor activity in patients with relapsed or refractory nasopharyngeal carcinoma.[27] (Refer to the PDQ summary on Nasopharyngeal Cancer Treatment for more information.) Treatment Options Under Clinical Evaluation The following is an example of a national and/or institutional clinical trial that is currently being conducted. Information about ongoing clinical trials is available from the NCI website. - H-25145 (NCT00953420) (Carboplatin, Docetaxel, and Laboratory-Treated T Cells in Treating Patients With Refractory or Relapsed EBV-Positive Nasopharyngeal Cancer): The objective of this trial is to determine the overall response rate in patients with relapsed/refractory, advanced-stage, EBV-positive nasopharyngeal carcinoma after treatment with docetaxel and carboplatin followed by immunotherapy with EBV-specific cytotoxic T lymphocytes. Individuals aged 10 years and older are eligible for this trial.
Esthesioneuroblastoma Incidence Esthesioneuroblastoma (olfactory neuroblastoma) is a small round-cell tumor arising from the nasal neuroepithelium that is distinct from primitive neuroectodermal tumors.[28,29,30,31] In children, esthesioneuroblastoma is a very rare malignancy, with an estimated incidence of 0.1 cases per 100,000 children younger than 15 years.[32] Despite its rarity, esthesioneuroblastoma is the most common cancer of the nasal cavity in pediatric patients, accounting for 28% of cases in a SEER study.[33] In a series of 511 patients from the SEER database, there was a slight male predominance, the mean age at presentation was 53 years, and only 8% of cases were younger than 25 years.[34] Most patients were white (81%) and the most common tumor sites were the nasal cavity (72%) and ethmoid sinus (13%).[34] Clinical Presentation Most children present in the second decade of life with symptoms that include the following: - Nasal obstruction.
- Epistaxis.
- Hyposmia.
- Exophthalmos.
- Nasopharyngeal mass, which may have local extension into the orbits, sinuses, or frontal lobe.
Staging Tumors are staged according to the Kadish system (refer to Table 1). Correlated with Kadish stage, prognosis ranges from 90% (stage A) to less than 40% (stage D). Most patients present with locally advanced-stage disease (Kadish stages B and C) and almost one-third of patients show tumor at distant sites (Kadish stage D).[32,33] Recent reports suggest that PET-CT may aid in staging the disease.[35] Table 1. Kadish Staging System| Stage | Description |
|---|
| A | Tumor confined to the nasal cavity. | | B | Tumor extending to the nasal sinuses. | | C | Tumor extending to the nasal sinuses and beyond. | | D | Tumor metastases present. | Review of multiple case series of mainly adult patients indicate that the following may correlate with adverse prognosis:[36,37,38] - Higher histopathologic grade.
- Positive surgical margin status.
- Metastases to the cervical lymph nodes.
Treatment and Outcome The use of multimodal therapy optimizes the chances for survival, with over 70% of children expected to survive 5 or more years after initial diagnosis.[32,39,40] A multi-institutional review of 24 patients younger than 21 years at diagnosis found a 5-year disease-free survival and OS of 73% to 74%.[41][Level of evidence: 3iiiA] Treatment options according to Kadish stage include the following:[42] - Kadish stage A: Surgery alone with clear margins. Adjuvant radiation therapy is indicated in patients with close and positive margins or with residual disease.
- Kadish stage B: Surgery followed by adjuvant radiation therapy. The role of adjuvant chemotherapy is controversial.
- Kadish stage C: Neoadjuvant approach with chemotherapy, radiation therapy, or concurrent chemotherapy-radiation therapy followed by surgery.
- Kadish stage D: Systemic chemotherapy and palliative radiation therapy to local and metastatic sites. Palliative care is incorporated into the treatment plan to improve quality of life.
The mainstay of treatment is surgery and radiation.[43] Newer techniques such as endoscopic sinus surgery may offer similar short-term outcomes to open craniofacial resection.[34]; [44][Level of evidence: 3iiiDii] Other techniques such as stereotactic radiosurgery and proton-beam therapy (charged-particle radiation therapy) may also play a role in the management of this tumor.[40,45] Nodal metastases are seen in about 5% of patients. Routine neck dissection and nodal exploration are not indicated in the absence of clinical or radiological evidence of disease.[46] Management of cervical lymph node metastases has been addressed in a review article.[46] Reports indicate promising results with the increased use of resection and neoadjuvant or adjuvant chemotherapy in patients with advanced-stage disease.[28,39,41,47,48]; [49][Level of evidence: 3iii] Chemotherapy regimens that have been used with efficacy include cisplatin with etoposide with or without ifosfamide;[42,50] vincristine, actinomycin D, and cyclophosphamide with or without doxorubicin; ifosfamide/etoposide; cisplatin plus etoposide or doxorubicin; [39] vincristine, doxorubicin, and cyclophosphamide;[51] and irinotecan plus docetaxel.[52][Level of evidence: 3iiA] Thyroid Tumors Incidence The annual incidence of thyroid cancers is 2.0 cases per 1 million people per year in children younger than 15 years, accounting for approximately 1.5% of all cancers in this age group.[3] Thyroid cancer incidence is higher in children aged 15 to 19 years (17.6 cases per 1 million people), and it accounts for approximately 8% of cancers arising in this older age group.[3,53] More thyroid carcinomas occur in females than in males.[54] A retrospective review of the SEER database from 1973 to 2011 identified 2,504 cases of papillary thyroid carcinoma in patients aged 20 years and younger.[53] The incidence of papillary thyroid carcinoma increased over this interval by roughly 2% each year. The trend toward larger tumors suggests that diagnostic scrutiny is not the only explanation for the observed results.[55] Risk Factors There is an excessive frequency of thyroid adenoma and carcinoma in patients who previously received radiation to the neck.[56,57] In the decade following the Chernobyl nuclear incident, there was a tenfold increase in the incidence of thyroid cancer compared with the previous and following decades.[58] In this group of patients with exposure to low-dose radiation, tumors commonly show a gain of 7q11.[59] When occurring in patients with the multiple endocrine neoplasia syndromes, thyroid cancer may be associated with the development of other types of malignant tumors. (Refer to the Multiple Endocrine Neoplasia (MEN) Syndromes and Carney Complex section of this summary for more information.) Histology Tumors of the thyroid are classified as adenomas or carcinomas.[60,61,62] Adenomas are benign, well circumscribed and encapsulated nodules that may cause enlargement of all or part of the gland, which extends to both sides of the neck and can be quite large; some tumors may secrete hormones. Transformation to a malignant carcinoma may occur in some cells, which may grow and spread to lymph nodes in the neck or to the lungs. Approximately 20% of thyroid nodules in children are malignant.[60,63] Various histologies account for the general diagnostic category of carcinoma of the thyroid; papillary and follicular carcinoma are often referred to as differentiated thyroid carcinoma:[57] - Papillary carcinoma (60%-75%): Papillary carcinoma often has a multicentric origin and a very high rate of lymph node metastasis (70%-90%).[64] Metastases to the lungs occur in about 25% of cases. Papillary carcinoma generally has a benign course, with a 10-year survival rate of more than 95%.[65,66] Overall, long-term outcomes for children and adolescents with papillary thyroid cancer are excellent, with 2% cause-specific mortality at 40 years.[66]
- Follicular carcinoma (10%-20%): Follicular carcinoma is usually encapsulated and has a higher incidence of bone and lung metastases.[64] It may be sporadic or familial.[67] Follicular carcinoma also has a generally benign course, with a 10-year survival rate of more than 95%.[65]
- Medullary carcinoma (5%-10%): Medullary carcinoma is a form of thyroid carcinoma that originates from the calcitonin-secreting parafollicular C cells. It is usually familial.[67]
- Anaplastic carcinoma (<1%).
Molecular Features and Tumor Characteristics Studies have shown subtle differences between the genetic profiling of childhood differentiated thyroid carcinomas and that of adult tumors. In one study, a higher prevalence of RET/PTC rearrangements was reported in pediatric papillary carcinoma (45%-65% in children vs. 3%-34% in adults).[68]BRAF V600E mutations are seen in more than 50% of adults with papillary thyroid carcinoma;[69] although it likely occurs in a similar frequency in pediatric patients, studies have revealed a wide variation in frequency of this mutation.[68,69,70,71] In children, the correlation between the genomic alteration and stage or prognosis has not been well defined. While two studies failed to show a correlation,[70,71] one study that included 55 pediatric thyroid carcinoma cases demonstrated a significant correlation between the presence of a BRAF V600E mutation and an increased risk of recurrence.[72] Differentiated thyroid carcinoma has been associated with germline DICER1 mutations and it is considered part of the DICER1 syndrome.[73] Table 2. Comparison of Thyroid Carcinoma Characteristics in Children and Adolescents and Adultsa| Characteristic | Children and Adolescents (%) | Adults (%) |
|---|
| a Yamashita et al.,[74]Nikita et al.,[71]and Alzahrani et al.[72] | | Histologic subtype: | | | | Papillary | 67-98 | 85-90 | | Follicular | 4-23 | <10 | | Medullary | 2-8 | 3 | | Poorly differentiated | <0.1 | 2-7 | | | | Gene rearrangements: | | | | RET/PTC | 21-87 | 0-35 | | NTRK 1 | 5-11 | 5-13 | | AKAP9-BRAF | 11 | 1 | | PAX8-PPARG | Unknown | 0-50 | | | | Point mutations: | | | | BRAF | 0-63 | 0-43 | | RASfamily | 0-16 | 25-69 | | GNAS | 0 | 11 | | TP53 | 0-23 | 0-20 | | TERT | 0 | 16 | | | | Other: | | | | Multicentric | 30-50 | 40-56 | | Lymph node involvement | 30-90 | 5-55 | | Extrathyroid extension | 24-51 | 16-46 | | Vascular invasion | <31 | 14-37 | | Distant metastases | 10-20 | 5-10 | Clinical Presentation and Outcome Patients with thyroid cancer usually present with a thyroid mass with or without painless cervical adenopathy.[75,76,77] On the basis of medical and family history and clinical constellation, the thyroid cancer may be part of a tumor predisposition syndrome such as MEN or DICER-1 syndrome.[78] Younger age is associated with a more aggressive clinical presentation in differentiated thyroid carcinoma. Children have a higher proportion of nodal involvement (40%-90% in children vs. 20%-50% in adults) and lung metastases (20%-30% in children vs. 2% in adults) than do adults.[69] Likewise, when compared with pubertal adolescents, prepubertal children have a more aggressive presentation with a greater degree of extrathyroid extension, lymph node involvement, and lung metastases. However, outcome is similar in the prepubertal and adolescent groups.[79] In well-differentiated thyroid cancer, male gender, large tumor size, and distant metastases have been found to have prognostic significance for early mortality; however, even patients in the highest risk group who have distant metastases had excellent survival at 90%.[80] A French registry analysis found similar outcomes in children and young adults who developed papillary thyroid carcinoma after previous radiation therapy compared with children and young adults who developed spontaneous papillary thyroid carcinoma; patients with previous thyroid irradiation for benign disease, however, presented with more invasive tumors and lymph node involvement.[81] Diagnostic Evaluation Initial evaluation of a child or adolescent with a thyroid nodule includes the following: - Ultrasound of the thyroid.
- Serum thyroid-stimulating hormone (TSH) level.
- Serum thyroglobulin level.
Tests of thyroid function are usually normal, but thyroglobulin can be elevated. Fine-needle aspiration as an initial diagnostic approach is sensitive and useful. However, in doubtful cases, open biopsy or resection should be considered.[82,83,84,85] Open biopsy or resection may also be preferable for young children. Table 3. Thyroid Carcinomas in Children| Histology | Associated Chromosomal Abnormality | Presentation | Diagnosis | Treatment |
|---|
| EGF = epidermal growth factor; MEN2 = multiple endocrine neoplasia type 2; TSH = thyroid-stimulating hormone. | | Differentiated thyroid carcinoma | RET/PTCmore common in children.BRAFV600E mutations occur with similar frequency in children and adults. Association with rare hereditary tumor syndromes:APC-associated polyposis,DICER1syndrome, Carney complex,PTENhamartoma tumor syndrome, Werner syndrome. | Thyroid mass. Prepubertal children more often with nodal and lung metastases. | Ultrasound, TSH, thyroglobulin. Fine needle or open biopsy. | Total or near-total thyroidectomy; I-131; thyroid hormone. In metastatic or recurrent disease, tyrosine kinase or EGF receptor inhibitors may be of benefit. | | Medullary thyroid carcinoma | MEN2 | Aggressive. 50% with metastases at presentation. | In familial MEN2,RETtesting. | Aggressive surgical intervention. Prophylactic thyroidectomy is indicated in familial cases. | Treatment of Papillary and Follicular (Differentiated) Thyroid Carcinoma Treatment options for papillary and follicular (differentiated) thyroid carcinoma may include the following: - Surgery.
- Radioactive iodine ablation.
The management of differentiated thyroid cancer in children has been reviewed in detail.[63,86] In 2015, the American Thyroid Association (ATA) Task Force on Pediatric Thyroid Cancer published guidelines for the management of thyroid nodules and differentiated thyroid cancer in children and adolescents. These guidelines (summarized below) are based on scientific evidence and expert panel opinion, with a careful assessment of the level of evidence.[63] - Preoperative evaluation.
- A comprehensive ultrasound of all regions of the neck using a high-resolution probe and Doppler technique should be obtained by an experienced ultrasonographer. A complete ultrasound examination should be performed before surgery.
- The addition of cross-sectional imaging (contrast-enhanced CT or magnetic resonance imaging) should be considered when there is concern about invasion of the aerodigestive tract. Importantly, if iodinated contrast agents are used, further evaluation and treatment with radioactive iodine may need to be delayed for 2 to 3 months until total body iodine burden decreases.
- Chest imaging (x-ray or CT) may be considered for patients with substantial cervical lymph node disease.
- Thyroid nuclear scintigraphy should be pursued only if the patient presents with a suppressed TSH.
- The routine use of bone scan or 2-deoxy-2-[18F]-fluoro-deoxyglucose (18 F-FDG) PET is not recommended.
- Surgery.
Pediatric thyroid surgery should ideally be performed by a surgeon who performs at least 30 or more cervical endocrine procedures annually in a hospital with the full spectrum of pediatric specialty care. - Thyroidectomy:
For patients with papillary or follicular carcinoma, total thyroidectomy is the recommended treatment of choice. The ATA expert panel recommendation is based on data showing an increased incidence of bilateral (30%) and multifocal (65%) disease. In patients with a small unilateral tumor confined to the gland, a near-total thyroidectomy-whereby a small amount of thyroid tissue (<1%-2%) is left in place at the entry point of the recurrent laryngeal nerve or superior parathyroid glands-might be considered to decrease permanent damage to those structures. Total thyroidectomy also optimizes the use of radioactive iodine for imaging and treatment. - Central neck dissection:
- A therapeutic central neck lymph node dissection should be done in the presence of clinical evidence of central or lateral neck metastases.
- For patients with no clinical evidence of gross extrathyroidal invasion or locoregional metastasis, a prophylactic central neck dissection should be considered on the basis of tumor focality and size of the primary tumor. For patients with unifocal disease, ipsilateral central neck dissection, with contralateral central neck dissection based on intraoperative findings, may also be considered.
- Lateral neck dissection:
- Cytological confirmation of metastatic disease to lymph nodes in the lateral neck is recommended before surgery.
- Routine prophylactic lateral neck dissection is not recommended.
- Classification and risk assignment.
Despite the limited data in pediatrics, the ATA Task Force recommends the use of the TNM classification system to categorize patients into one of three risk groups. (Refer to the Stage Information for Thyroid Cancer section in the PDQ summary on Thyroid Cancer Treatment for more information about the TNM system.) This categorization strategy is meant to define the risk of persistent cervical disease and help determine which patients should undergo postoperative staging for the presence of distant metastasis. - ATA Pediatric Low Risk: Disease confined to the thyroid with N0 or NX disease or patients with incidental N1a (microscopic metastasis to a small number of central neck nodes). These patients are at lowest risk of distant disease but may still be at risk of residual cervical disease, especially if the initial surgery did not include central neck dissection.
- ATA Pediatric Intermediate Risk: Extensive N1a or minimal N1b disease. These patients are at low risk of distant metastasis but are at an increased risk of incomplete lymph node resection and persistent cervical disease.
- ATA Pediatric High Risk: Regionally extensive disease (N1b) or locally invasive disease (T4), with or without distant metastasis. Patients in this group are at the highest risk of incomplete resection, persistent disease, and distant metastasis.
- Postoperative staging.
Initial staging should be performed within 12 weeks after surgery; the purpose is to assess for evidence of persistent locoregional disease and to identify patients who are likely to benefit from additional therapy with 131 I. The ATA Pediatric Risk Level (as defined above) helps determine the extent of postoperative testing. - ATA Pediatric Low Risk: Initial postoperative staging includes a TSH-suppressed thyroglobulin
- ATA Pediatric Intermediate and High Risk: TSH-stimulated thyroglobulin and diagnostic 123 I whole-body scan are recommended for further stratification and determination with 131 I.
For patients with anti-thyroglobulin antibodies, consideration can be given to deferred postoperative staging to allow time for antibody clearance, except in patients with T4 or M1 disease. - Radioactive iodine ablation.
The goal of 131 I therapy is to decrease the risks of recurrence and to decrease mortality by eliminating iodine-avid disease. - The ATA Task Force recommends the use of 131 I for the treatment of iodine-avid persistent locoregional or nodal disease that cannot be resected and known or presumed iodine-avid distant metastases. For patients with persistent disease after administration of 131 I, the decision to pursue additional 131 I therapy should be individualized on the basis of clinical data and previous response.
- To facilitate 131 I uptake by residual iodine-avid disease, the TSH level should be above 30 mIU/L. This level can be achieved by withdrawing levothyroxine for at least 14 days. In patients who cannot mount an adequate TSH response or cannot tolerate profound hypothyroidism, recombinant human TSH may be used.
- Therapeutic 131 I administration is commonly based on either empiric dosing or whole-body dosimetry. Based on the lack of data comparing empiric treatment and treatment informed by dosimetry, the ATA Task Force was unable to recommend one specific approach. However, because of the differences in body size and iodine clearance in children compared with adults, it is recommended that all activities of 131 I should be calculated by experts with experience in dosing children.
- A posttreatment whole-body scan is recommended for all children 4 to 7 days after 131 I therapy. The addition of single-photon emission CT with integrated conventional CT (SPECT/CT) may help to distinguish the anatomic location of focal uptake.
- Surveillance and follow-up.
The ATA Task Force recommends the following risk-adapted approach to follow-up: - ATA Pediatric Low Risk:
- TSH goal of 0.5 to 1.0 mIU/L.
- Ultrasound at 6 months and then annually for 5 years.
- Thyroglobulin levels on levothyroxine treatment every 3 to 6 months for 2 years and then annually.
- ATA Pediatric Intermediate Risk:
- TSH goal of 0.1 to 0.5 mIU/L. In patients who have no evidence of disease after 3 to 5 years of follow-up, the TSH can be allowed to rise to the low-normal range.
- Ultrasound at 6 months postoperatively, every 6 to 12 months for 5 years, and then less frequently.
- Thyroglobulin levels on levothyroxine treatment every 3 to 6 months for 3 years and then annually.
- Consider TSH-stimulated thyroglobulin and diagnostic 123 I scan in 1 to 2 years in patients treated with 131 I.
- ATA Pediatric High Risk:
- TSH goal of less than 0.1 mIU/L. In patients who have no evidence of disease after 3 to 5 years of follow-up, the TSH can be allowed to rise to the low-normal range.
- Ultrasound at 6 months postoperatively, every 6 to 12 months for 5 years, and then less frequently.
- Thyroglobulin levels on levothyroxine treatment every 3 to 6 months for 3 years and then annually.
- Consider TSH-stimulated thyroglobulin and diagnostic 123 I scan in 1 to 2 years in patients treated with 131 I.
Treatment of Recurrent Papillary and Follicular (Differentiated) Thyroid Carcinoma Patients with differentiated thyroid cancer generally have an excellent survival with relatively few side effects.[87,88,89] However, recurrence is common (35%-45%) and is seen more often in children younger than 10 years and in those with palpable cervical lymph nodes at diagnosis.[90,91] Even patients with a tumor that has spread to the lungs may expect to have no decrease in life span after appropriate treatment.[92] Of note, the sodium-iodide symporter (a membrane-bound glycoprotein cotransporter), essential for uptake of iodide and thyroid hormone synthesis, is expressed in 35% to 45% of thyroid cancers in children and adolescents. Patients with expression of the sodium-iodide symporter have a lower risk of recurrence.[93] Recurrent papillary thyroid cancer is usually responsive to treatment with radioactive iodine ablation.[94] TKIs such as sorafenib have been shown to induce responses in up to 15% of adult patients with metastatic disease.[95] Response to sorafenib has also been documented in a pediatric case.[96] TKIs approved for the treatment of adults include the following: - Sorafenib. Sorafenib is a vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and RAS kinase inhibitor. In a randomized phase III trial, sorafenib improved progression-free survival (PFS) when compared with placebo in adult patients with radioactive iodine-refractory locally advanced or metastatic differentiated thyroid cancer.[97] In one case report, sorafenib produced a radiographic response in a patient aged 8 years with metastatic papillary thyroid carcinoma.[98] Sorafenib was approved by the U.S. Food and Drug Administration (FDA) in November 2013 for the treatment of adults with late stage metastatic differentiated thyroid carcinoma.
- Lenvatinib. Lenvatinib is an oral VEGFR, fibroblast growth factor receptor, PDGFR, RET, and KIT inhibitor. In a phase III randomized study of adults with I-131 refractory differentiated thyroid cancer, lenvatinib was associated with a significant improvement in PFS and response rate when compared with a placebo.[99] Lenvatinib was approved by the FDA in February 2015 for the treatment of adults with progressive radioactive iodine-refractory differentiated thyroid carcinoma.
Given the high incidence of BRAF mutations in patients with papillary thyroid carcinoma, the use of selective RAF/MEK inhibitors is being investigated.[95,100,101] (Refer to the PDQ summary on adult Thyroid Cancer Treatment for more information.) Treatment of Medullary Thyroid Carcinoma Medullary thyroid carcinomas are commonly associated with the MEN2 syndrome (refer to the Multiple Endocrine Neoplasia (MEN) Syndromes and Carney Complex section of this summary for more information). They present with a more aggressive clinical course; 50% of the cases have hematogenous metastases at diagnosis.[102] Patients with medullary carcinoma of the thyroid have a guarded prognosis, unless they have very small tumors (microcarcinoma, defined as <1.0 cm in diameter), which carry a good prognosis.[103] A natural history study of children and young adults with medullary thyroid cancer is being conducted by the National Cancer Institute (NCT01660984). For patients with de novo RET mutations and no familial history, nonendocrine manifestations such as intestinal ganglioneuromatosis or skeletal or ocular stigmata, may facilitate early diagnosis and result in better outcomes.[104] Treatment options for medullary thyroid carcinoma include the following: - Surgery: Treatment for children with medullary thyroid carcinoma is mainly surgical. A review of 430 patients aged 0 to 21 years with medullary thyroid cancer reported that older age (16-21 years) at diagnosis, tumor diameter greater than 2 cm, positive margins after total thyroidectomy, and lymph node metastases were associated with a worse prognosis.[105] This suggests that central neck node dissection and dissection of nearby positive nodes should improve the 10-year survival for these patients.
Most cases of medullary thyroid carcinoma occur in the context of the MEN 2A and MEN 2B syndromes. In those familial cases, early genetic testing and counseling is indicated, and prophylactic surgery is recommended for children with the RET germline mutation. Strong genotype-phenotype correlations have facilitated the development of guidelines for intervention, including screening and age at which prophylactic thyroidectomy should occur.[102] - TKI therapy: A number of TKIs have been evaluated and approved for patients with advanced thyroid carcinoma.
- Vandetanib. Vandetanib (an inhibitor of RET kinase, VEGFR, and epidermal growth factor receptor signaling) is approved by the U.S. FDA for the treatment of symptomatic or progressive medullary thyroid cancer in adult patients with unresectable, locally advanced, or metastatic disease. Approval was based on a randomized, placebo-controlled, phase III trial that showed a marked PFS improvement for patients randomly assigned to receive vandetanib (hazard ratio, 0.35); the trial also showed an objective response rate advantage for patients receiving vandetanib (44% vs. 1% for the placebo arm).[106,107]
Children with locally advanced or metastatic medullary thyroid carcinoma were treated with vandetanib in a phase I/II trial. Of 16 patients, only 1 had no response, and 7 had a partial response, for an objective response rate of 44%. Disease in three of those patients subsequently recurred, but 11 of 16 patients treated with vandetanib remained on therapy at the time of the report. The median duration of therapy for the entire cohort was 27 months, with a range of 2 to 52 months.[108] - Cabozantinib. Cabozantinib (an inhibitor of the RET and MET kinases and VEGFR) has also shown activity against unresectable medullary thyroid cancer (10 of 35 adult patients [29%] had a partial response).[109] Cabozantinib was approved by the FDA in November 2012 for the treatment of adults with metastatic medullary thyroid cancer.
(Refer to the Multiple Endocrine Neoplasia (MEN) Syndromes and Carney Complex section of this summary and the Treatment for those with MTC section in the PDQ summary on Genetics of Endocrine and Neuroendocrine Neoplasias for more information.) Oral Cavity Cancer Incidence More than 90% of tumors and tumor-like lesions in the oral cavity are benign.[110,111,112,113] Cancer of the oral cavity is extremely rare in children and adolescents.[114,115] According to the SEER Stat Fact Sheets, only 0.6% of all cases are diagnosed in patients younger than 20 years, and in 2008, the age-adjusted incidence for this population was 0.24 cases per 100,000. The incidence of cancer of the oral cavity and pharynx has increased in adolescent and young adult females, and this pattern is consistent with the national increase in orogenital sexual intercourse in younger females and human papillomavirus (HPV) infection.[116] It is currently estimated that the prevalence of oral HPV infection in the United States is 6.9% in people aged 14 to 69 years and that HPV causes about 30,000 oropharyngeal cancers. Furthermore, from 1999 to 2008, the incidence rates for HPV-related oropharyngeal cancer have increased by 4.4% per year in white men and 1.9% in white women.[117,118,119] Current practices to increase HPV immunization rates in both boys and girls may reduce the burden of HPV-related cancers.[120] Histology Benign odontogenic neoplasms of the oral cavity include odontoma and ameloblastoma. The most common nonodontogenic neoplasms of the oral cavity are fibromas, hemangiomas, and papillomas. Tumor-like lesions of the oral cavity include lymphangiomas, granulomas, and Langerhans cell histiocytosis.[110,111,112,113] (Refer to the Oral cavity subsection in the PDQ summary on Langerhans Cell Histiocytosis Treatment for more information about Langerhans cell histiocytosis of the oral cavity.) Malignant lesions of the oral cavity were found in 0.1% to 2% of a series of oral biopsies performed in children [110,111] and 3% to 13% of oral tumor biopsies.[112,113] Malignant tumor types include lymphomas (especially Burkitt) and sarcomas (including rhabdomyosarcoma and fibrosarcoma). Mucoepidermoid carcinomas of the oral cavity have rarely been reported in the pediatric and adolescent age group. Most are low grade and have a high cure rate with surgery alone.[121]; [122][Level of evidence: 3iiiA] The most common type of primary oral cavity cancer in adults, squamous cell carcinoma (SCC), is extremely rare in children. Review of the SEER database identified 54 patients younger than 20 years with oral cavity SCC between 1973 and 2006. Pediatric patients with oral cavity SCC were more often female and had better survival than adult patients. When differences in patient, tumor, and treatment-related characteristics are adjusted for, the two groups experienced equivalent survival.[121][Level of evidence: 3iA] Diseases that can be associated with the development of oral cavity and/or head and neck SCC include Fanconi anemia, dyskeratosis congenita, connexin mutations, chronic graft-versus-host disease, epidermolysis bullosa, xeroderma pigmentosum, and HPV infection.[123,124,125,126,127,128,129,130] Treatment Treatment of benign oral cavity tumors is surgical. Management of malignant tumors of the oral cavity is dependent on histology and may include surgery, chemotherapy, and radiation.[131] Most reported cases of oral cavity SCC managed with surgery alone have done well without recurrence.[121,132] (Refer to the PDQ summary on adult Lip and Oral Cavity Cancer Treatment for more information.) Langerhans cell histiocytosis of the oral cavity may require treatment in addition to surgery. (Refer to the PDQ summary on Langerhans Cell Histiocytosis Treatment for more information.) Salivary Gland Tumors Incidence and Outcome Salivary gland tumors are rare and account for 0.5% of all malignancies in children and adolescents. After rhabdomyosarcoma, they are the most common tumor in the head and neck.[133,134] Salivary gland tumors may occur after radiation therapy and chemotherapy are given for treatment of primary leukemia or solid tumors.[135,136,137] Overall 5-year survival in the pediatric age group is approximately 95%.[138] A review of the SEER database identified 284 patients younger than 20 years with tumors of the parotid gland.[139][Level of evidence: 3iA] OS was 96% at 5 years, 95% at 10 years, and 83% at 20 years. Adolescents had higher mortality rates (7.1% vs. 1.6% for children aged <15 years, P = .23). Clinical Presentation Most salivary gland neoplasms arise in the parotid gland.[140,141,142,143,144,145,146] About 15% of these tumors arise in the submandibular glands or in the minor salivary glands under the tongue and jaw.[144] These tumors are most frequently benign but may be malignant, especially in young children.[147] Histology The most common malignant salivary gland tumor in children is mucoepidermoid carcinoma, followed by acinic cell carcinoma, and adenoid cystic carcinoma; less common malignancies include rhabdomyosarcoma, adenocarcinoma, and undifferentiated carcinoma.[133,144,146,148,149,150] Mucoepidermoid carcinoma is usually low or intermediate grade, although high-grade tumors occur. In one study, 12 of 12 tumors were positive for MECT1/MAML2 fusion transcripts. This reflects the common chromosome translocation t(11;19)(q21;p13) that is seen in adults with salivary gland tumors.[151] Mucoepidermoid carcinoma is the most common type of treatment-related salivary gland tumor, and with standard therapy, the 5-year survival is about 95%.[146,150,152,153] Treatment Radical surgical removal is the treatment of choice for salivary gland tumors whenever possible, with additional use of radiation therapy for high-grade tumors or tumors that have invasive characteristics, such as lymph node metastasis, lymphovascular invasion, or perineural extension.[138,149,154]; [145][Level of evidence: 3iiiA] One retrospective study compared proton and conventional radiation therapy and found proton therapy to have a favorable acute toxicity and dosimetric profile.[155] There are inadequate data regarding the efficacy of adjuvant chemotherapy in children. (Refer to the PDQ summary on adult Salivary Gland Cancer Treatment for more information.) Sialoblastoma Sialoblastoma is a usually benign tumor presenting in the neonatal period and rarely metastasizes.[156] Chemotherapy regimens with carboplatin, epirubicin, vincristine, etoposide, dactinomycin, doxorubicin, and ifosfamide have produced responses in two children with sialoblastoma.[157]; [158][Level of evidence: 3iiiDiv] Laryngeal Cancer and Papillomatosis Tumors of the larynx are rare. The most common benign tumor is subglottic hemangioma.[159] Malignant tumors, which are especially rare, may be associated with benign tumors such as polyps and papillomas.[160,161] These tumors may cause hoarseness, difficulty swallowing, and enlargement of the lymph nodes of the neck. Treatment of Laryngeal Cancer Rhabdomyosarcoma is the most common malignant tumor of the larynx in the pediatric age group and is treated with chemotherapy and radiation therapy.[162] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.) SCC of the larynx is managed in the same manner as in adults with carcinoma at this site, with surgery and radiation.[163] Laser surgery may be the initial treatment utilized for these lesions. (Refer to the PDQ summary on Laryngeal Cancer Treatment for more information about treatment of laryngeal cancer in adults.) Treatment of Papillomatosis Recurrent respiratory papillomatosis is the most common benign laryngeal tumor in children and is associated with HPV infection, most commonly HPV-6 and HPV-11.[164] The presence of HPV-11 appears to correlate with a more aggressive clinical course than HPV-6.[165] These tumors can cause hoarseness because of their association with wart-like nodules on the vocal cords and may rarely extend into the lung, producing significant morbidity.[166] Malignant degeneration may occur with development of cancer in the larynx and squamous cell lung cancer. Papillomatosis is not cancerous, and primary treatment is surgical ablation with laser vaporization.[167] Frequent recurrences are common. Lung involvement, although rare, can occur.[166] If a patient requires more than four surgical procedures per year, other interventions may be necessary, including the following: - Interferon.[168]
- Immunotherapy with HspE7, a recombinant fusion protein that has shown activity in other HPV-related diseases. A pilot study suggested a marked increase in the amount of time between surgeries.[169]
- Laser therapy combined with intralesional bevacizumab.[170]
The effectiveness of intralesional cidofovir has not been conclusively demonstrated.[171] Midline Tract Carcinoma Involving theNUTGene (NUTMidline Carcinoma) NUT midline carcinoma is a very rare and aggressive malignancy genetically defined by rearrangements of the gene NUT. In the majority (75%) of cases, the NUT gene on chromosome 15q14 is fused with BRD4 on chromosome 19p13, creating chimeric genes that encode the BRD-NUT fusion proteins. In the remaining cases, NUT is fused to BRD3 on chromosome 9q34 or to NSD3 on chromosome 8p11;[172] these tumors are termed NUT-variant.[173] The tumors arise in midline epithelial structures, typically mediastinum and upper aerodigestive tract, and present as very aggressive undifferentiated carcinomas, with or without squamous differentiation.[174] Although the original description of this neoplasm was made in children and young adults, individuals of all ages can be affected.[173] A retrospective series with clinicopathologic correlation found that the median age at diagnosis of 54 patients was 16 years (range, 0.1-78 years).[175] The outcome is very poor, with an average survival of less than 1 year. Preliminary data seem to indicate that NUT-variant tumors may have a more protracted course.[173,174] Treatment Treatment includes a multimodal approach with systemic chemotherapy, surgery, and radiation therapy. Cisplatin, taxanes, and alkylating agents have been used with some success; however, while early response is common, tumors progress early in the course of the disease.[175][Level of evidence: 3iiiB] Preclinical studies have shown that NUT-BRD4 is associated with globally decreased histone acetylation and transcriptional repression; studies have also shown that this acetylation can be restored with histone deacetylase inhibitors, resulting in squamous differentiation and arrested growth in vitro and growth inhibition in xenograft models. Response to vorinostat has been reported in two separate cases of children with refractory disease, thus suggesting a potential role for this class of agents in the treatment of this malignancy.[176,177] The BET bromodomain inhibitors represent a promising class of agents that is being investigated for adults with this malignancy.[172] Treatment Options Under Clinical Evaluation The following are examples of national and/or institutional clinical trials that are currently being conducted. Information about ongoing clinical trials is available from the NCI website. - NCT01587703(A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Activity of GSK525762 in Subjects With NUT Midline Carcinoma and Other Cancers): This study is evaluating the safety, pharmacokinetic, and pharmacodynamic profiles observed after oral administration of GSK525762, a BET bromodomain inhibitor, as well as the tolerability and clinical activity, in patients with NUT midline carcinoma and other solid tumors. Patients aged 16 years and older are eligible for this study.
- NCT01987362 (A Two Part, Multicenter, Open-label Study of TEN-010 Given Subcutaneously): This is a phase I, nonrandomized, dose-escalating, open label, multicenter study of patients aged 18 years or older with histologically confirmed advanced solid tumors with progressive disease requiring therapy or NUT midline carcinoma. This study is evaluating the safety, tolerability, and pharmacokinetics of TEN-010, a small molecule bromodomain inhibitor.
References:
-
Ayan I, Kaytan E, Ayan N: Childhood nasopharyngeal carcinoma: from biology to treatment. Lancet Oncol 4 (1): 13-21, 2003.
-
Yan Z, Xia L, Huang Y, et al.: Nasopharyngeal carcinoma in children and adolescents in an endemic area: a report of 185 cases. Int J Pediatr Otorhinolaryngol 77 (9): 1454-60, 2013.
-
Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. Bethesda, Md: National Cancer Institute, 2009. Also available online. Last accessed April 04, 2017.
-
Sultan I, Casanova M, Ferrari A, et al.: Differential features of nasopharyngeal carcinoma in children and adults: a SEER study. Pediatr Blood Cancer 55 (2): 279-84, 2010.
-
Richards MK, Dahl JP, Gow K, et al.: Factors Associated With Mortality in Pediatric vs Adult Nasopharyngeal Carcinoma. JAMA Otolaryngol Head Neck Surg 142 (3): 217-22, 2016.
-
Dawson CW, Port RJ, Young LS: The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin Cancer Biol 22 (2): 144-53, 2012.
-
Lo YM, Chan LY, Lo KW, et al.: Quantitative analysis of cell-free Epstein-Barr virus DNA in plasma of patients with nasopharyngeal carcinoma. Cancer Res 59 (6): 1188-91, 1999.
-
Hu S, Xu X, Xu J, et al.: Prognostic factors and long-term outcomes of nasopharyngeal carcinoma in children and adolescents. Pediatr Blood Cancer 60 (7): 1122-7, 2013.
-
Cheuk DK, Sabin ND, Hossain M, et al.: PET/CT for staging and follow-up of pediatric nasopharyngeal carcinoma. Eur J Nucl Med Mol Imaging 39 (7): 1097-106, 2012.
-
Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010.
-
Casanova M, Ferrari A, Gandola L, et al.: Undifferentiated nasopharyngeal carcinoma in children and adolescents: comparison between staging systems. Ann Oncol 12 (8): 1157-62, 2001.
-
Cheuk DK, Billups CA, Martin MG, et al.: Prognostic factors and long-term outcomes of childhood nasopharyngeal carcinoma. Cancer 117 (1): 197-206, 2011.
-
Casanova M, Bisogno G, Gandola L, et al.: A prospective protocol for nasopharyngeal carcinoma in children and adolescents: the Italian Rare Tumors in Pediatric Age (TREP) project. Cancer 118 (10): 2718-25, 2012.
-
Buehrlen M, Zwaan CM, Granzen B, et al.: Multimodal treatment, including interferon beta, of nasopharyngeal carcinoma in children and young adults: preliminary results from the prospective, multicenter study NPC-2003-GPOH/DCOG. Cancer 118 (19): 4892-900, 2012.
-
Sahai P, Mohanti BK, Sharma A, et al.: Clinical outcome and morbidity in pediatric patients with nasopharyngeal cancer treated with chemoradiotherapy. Pediatr Blood Cancer 64 (2): 259-266, 2017.
-
Al-Sarraf M, LeBlanc M, Giri PG, et al.: Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized Intergroup study 0099. J Clin Oncol 16 (4): 1310-7, 1998.
-
Wolden SL, Steinherz PG, Kraus DH, et al.: Improved long-term survival with combined modality therapy for pediatric nasopharynx cancer. Int J Radiat Oncol Biol Phys 46 (4): 859-64, 2000.
-
Langendijk JA, Leemans ChR, Buter J, et al.: The additional value of chemotherapy to radiotherapy in locally advanced nasopharyngeal carcinoma: a meta-analysis of the published literature. J Clin Oncol 22 (22): 4604-12, 2004.
-
Venkitaraman R, Ramanan SG, Sagar TG: Nasopharyngeal cancer of childhood and adolescence: a single institution experience. Pediatr Hematol Oncol 24 (7): 493-502, 2007 Oct-Nov.
-
Yan M, Kumachev A, Siu LL, et al.: Chemoradiotherapy regimens for locoregionally advanced nasopharyngeal carcinoma: A Bayesian network meta-analysis. Eur J Cancer 51 (12): 1570-9, 2015.
-
Mertens R, Granzen B, Lassay L, et al.: Treatment of nasopharyngeal carcinoma in children and adolescents: definitive results of a multicenter study (NPC-91-GPOH). Cancer 104 (5): 1083-9, 2005.
-
Rodriguez-Galindo C, Wofford M, Castleberry RP, et al.: Preradiation chemotherapy with methotrexate, cisplatin, 5-fluorouracil, and leucovorin for pediatric nasopharyngeal carcinoma. Cancer 103 (4): 850-7, 2005.
-
Casanova M, Özyar E, Patte C, et al.: International randomized phase 2 study on the addition of docetaxel to the combination of cisplatin and 5-fluorouracil in the induction treatment for nasopharyngeal carcinoma in children and adolescents. Cancer Chemother Pharmacol 77 (2): 289-98, 2016.
-
Nakamura RA, Novaes PE, Antoneli CB, et al.: High-dose-rate brachytherapy as part of a multidisciplinary treatment of nasopharyngeal lymphoepithelioma in childhood. Cancer 104 (3): 525-31, 2005.
-
Louis CU, Paulino AC, Gottschalk S, et al.: A single institution experience with pediatric nasopharyngeal carcinoma: high incidence of toxicity associated with platinum-based chemotherapy plus IMRT. J Pediatr Hematol Oncol 29 (7): 500-5, 2007.
-
Varan A, Ozyar E, Corapçioğlu F, et al.: Pediatric and young adult nasopharyngeal carcinoma patients treated with preradiation Cisplatin and docetaxel chemotherapy. Int J Radiat Oncol Biol Phys 73 (4): 1116-20, 2009.
-
Straathof KC, Bollard CM, Popat U, et al.: Treatment of nasopharyngeal carcinoma with Epstein-Barr virus--specific T lymphocytes. Blood 105 (5): 1898-904, 2005.
-
Kumar M, Fallon RJ, Hill JS, et al.: Esthesioneuroblastoma in children. J Pediatr Hematol Oncol 24 (6): 482-7, 2002 Aug-Sep.
-
Theilgaard SA, Buchwald C, Ingeholm P, et al.: Esthesioneuroblastoma: a Danish demographic study of 40 patients registered between 1978 and 2000. Acta Otolaryngol 123 (3): 433-9, 2003.
-
Dias FL, Sa GM, Lima RA, et al.: Patterns of failure and outcome in esthesioneuroblastoma. Arch Otolaryngol Head Neck Surg 129 (11): 1186-92, 2003.
-
Nakao K, Watanabe K, Fujishiro Y, et al.: Olfactory neuroblastoma: long-term clinical outcome at a single institute between 1979 and 2003. Acta Otolaryngol Suppl (559): 113-7, 2007.
-
Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
-
Benoit MM, Bhattacharyya N, Faquin W, et al.: Cancer of the nasal cavity in the pediatric population. Pediatrics 121 (1): e141-5, 2008.
-
Soler ZM, Smith TL: Endoscopic versus open craniofacial resection of esthesioneuroblastoma: what is the evidence? Laryngoscope 122 (2): 244-5, 2012.
-
Broski SM, Hunt CH, Johnson GB, et al.: The added value of 18F-FDG PET/CT for evaluation of patients with esthesioneuroblastoma. J Nucl Med 53 (8): 1200-6, 2012.
-
Dulguerov P, Allal AS, Calcaterra TC: Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2 (11): 683-90, 2001.
-
Patel SG, Singh B, Stambuk HE, et al.: Craniofacial surgery for esthesioneuroblastoma: report of an international collaborative study. J Neurol Surg B Skull Base 73 (3): 208-20, 2012.
-
Herr MW, Sethi RK, Meier JC, et al.: Esthesioneuroblastoma: an update on the massachusetts eye and ear infirmary and massachusetts general hospital experience with craniofacial resection, proton beam radiation, and chemotherapy. J Neurol Surg B Skull Base 75 (1): 58-64, 2014.
-
Eich HT, Müller RP, Micke O, et al.: Esthesioneuroblastoma in childhood and adolescence. Better prognosis with multimodal treatment? Strahlenther Onkol 181 (6): 378-84, 2005.
-
Lucas JT Jr, Ladra MM, MacDonald SM, et al.: Proton therapy for pediatric and adolescent esthesioneuroblastoma. Pediatr Blood Cancer 62 (9): 1523-8, 2015.
-
Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
-
Kumar R: Esthesioneuroblastoma: Multimodal management and review of literature. World J Clin Cases 3 (9): 774-8, 2015.
-
Ozsahin M, Gruber G, Olszyk O, et al.: Outcome and prognostic factors in olfactory neuroblastoma: a rare cancer network study. Int J Radiat Oncol Biol Phys 78 (4): 992-7, 2010.
-
Gallia GL, Reh DD, Lane AP, et al.: Endoscopic resection of esthesioneuroblastoma. J Clin Neurosci 19 (11): 1478-82, 2012.
-
Unger F, Haselsberger K, Walch C, et al.: Combined endoscopic surgery and radiosurgery as treatment modality for olfactory neuroblastoma (esthesioneuroblastoma). Acta Neurochir (Wien) 147 (6): 595-601; discussion 601-2, 2005.
-
Zanation AM, Ferlito A, Rinaldo A, et al.: When, how and why to treat the neck in patients with esthesioneuroblastoma: a review. Eur Arch Otorhinolaryngol 267 (11): 1667-71, 2010.
-
Loy AH, Reibel JF, Read PW, et al.: Esthesioneuroblastoma: continued follow-up of a single institution's experience. Arch Otolaryngol Head Neck Surg 132 (2): 134-8, 2006.
-
Porter AB, Bernold DM, Giannini C, et al.: Retrospective review of adjuvant chemotherapy for esthesioneuroblastoma. J Neurooncol 90 (2): 201-4, 2008.
-
Benfari G, Fusconi M, Ciofalo A, et al.: Radiotherapy alone for local tumour control in esthesioneuroblastoma. Acta Otorhinolaryngol Ital 28 (6): 292-7, 2008.
-
Kim DW, Jo YH, Kim JH, et al.: Neoadjuvant etoposide, ifosfamide, and cisplatin for the treatment of olfactory neuroblastoma. Cancer 101 (10): 2257-60, 2004.
-
El Kababri M, Habrand JL, Valteau-Couanet D, et al.: Esthesioneuroblastoma in children and adolescent: experience on 11 cases with literature review. J Pediatr Hematol Oncol 36 (2): 91-5, 2014.
-
Kiyota N, Tahara M, Fujii S, et al.: Nonplatinum-based chemotherapy with irinotecan plus docetaxel for advanced or metastatic olfactory neuroblastoma: a retrospective analysis of 12 cases. Cancer 112 (4): 885-91, 2008.
-
Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
-
Shapiro NL, Bhattacharyya N: Population-based outcomes for pediatric thyroid carcinoma. Laryngoscope 115 (2): 337-40, 2005.
-
Vergamini LB, Frazier AL, Abrantes FL, et al.: Increase in the incidence of differentiated thyroid carcinoma in children, adolescents, and young adults: a population-based study. J Pediatr 164 (6): 1481-5, 2014.
-
Cotterill SJ, Pearce MS, Parker L: Thyroid cancer in children and young adults in the North of England. Is increasing incidence related to the Chernobyl accident? Eur J Cancer 37 (8): 1020-6, 2001.
-
Kaplan MM, Garnick MB, Gelber R, et al.: Risk factors for thyroid abnormalities after neck irradiation for childhood cancer. Am J Med 74 (2): 272-80, 1983.
-
Demidchik YE, Saenko VA, Yamashita S: Childhood thyroid cancer in Belarus, Russia, and Ukraine after Chernobyl and at present. Arq Bras Endocrinol Metabol 51 (5): 748-62, 2007.
-
Hess J, Thomas G, Braselmann H, et al.: Gain of chromosome band 7q11 in papillary thyroid carcinomas of young patients is associated with exposure to low-dose irradiation. Proc Natl Acad Sci U S A 108 (23): 9595-600, 2011.
-
Dinauer C, Francis GL: Thyroid cancer in children. Endocrinol Metab Clin North Am 36 (3): 779-806, vii, 2007.
-
Vasko V, Bauer AJ, Tuttle RM, et al.: Papillary and follicular thyroid cancers in children. Endocr Dev 10: 140-72, 2007.
-
Halac I, Zimmerman D: Thyroid nodules and cancers in children. Endocrinol Metab Clin North Am 34 (3): 725-44, x, 2005.
-
Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
-
Feinmesser R, Lubin E, Segal K, et al.: Carcinoma of the thyroid in children--a review. J Pediatr Endocrinol Metab 10 (6): 561-8, 1997 Nov-Dec.
-
Hung W, Sarlis NJ: Current controversies in the management of pediatric patients with well-differentiated nonmedullary thyroid cancer: a review. Thyroid 12 (8): 683-702, 2002.
-
Hay ID, Gonzalez-Losada T, Reinalda MS, et al.: Long-term outcome in 215 children and adolescents with papillary thyroid cancer treated during 1940 through 2008. World J Surg 34 (6): 1192-202, 2010.
-
Skinner MA: Management of hereditary thyroid cancer in children. Surg Oncol 12 (2): 101-4, 2003.
-
Ballester LY, Sarabia SF, Sayeed H, et al.: Integrating Molecular Testing in the Diagnosis and Management of Children with Thyroid Lesions. Pediatr Dev Pathol 19 (2): 94-100, 2016 Mar-Apr.
-
Rivkees SA, Mazzaferri EL, Verburg FA, et al.: The treatment of differentiated thyroid cancer in children: emphasis on surgical approach and radioactive iodine therapy. Endocr Rev 32 (6): 798-826, 2011.
-
Henke LE, Perkins SM, Pfeifer JD, et al.: BRAF V600E mutational status in pediatric thyroid cancer. Pediatr Blood Cancer 61 (7): 1168-72, 2014.
-
Nikita ME, Jiang W, Cheng SM, et al.: Mutational Analysis in Pediatric Thyroid Cancer and Correlations with Age, Ethnicity, and Clinical Presentation. Thyroid 26 (2): 227-34, 2016.
-
Alzahrani AS, Qasem E, Murugan AK, et al.: Uncommon TERT Promoter Mutations in Pediatric Thyroid Cancer. Thyroid 26 (2): 235-41, 2016.
-
Slade I, Bacchelli C, Davies H, et al.: DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 48 (4): 273-8, 2011.
-
Yamashita S, Saenko V: Mechanisms of Disease: molecular genetics of childhood thyroid cancers. Nat Clin Pract Endocrinol Metab 3 (5): 422-9, 2007.
-
Thompson GB, Hay ID: Current strategies for surgical management and adjuvant treatment of childhood papillary thyroid carcinoma. World J Surg 28 (12): 1187-98, 2004.
-
Rachmiel M, Charron M, Gupta A, et al.: Evidence-based review of treatment and follow up of pediatric patients with differentiated thyroid carcinoma. J Pediatr Endocrinol Metab 19 (12): 1377-93, 2006.
-
Wada N, Sugino K, Mimura T, et al.: Treatment strategy of papillary thyroid carcinoma in children and adolescents: clinical significance of the initial nodal manifestation. Ann Surg Oncol 16 (12): 3442-9, 2009.
-
Schultz KA, Yang J, Doros L, et al.: DICER1-pleuropulmonary blastoma familial tumor predisposition syndrome: a unique constellation of neoplastic conditions. Pathol Case Rev 19 (2): 90-100, 2014.
-
Lazar L, Lebenthal Y, Steinmetz A, et al.: Differentiated thyroid carcinoma in pediatric patients: comparison of presentation and course between pre-pubertal children and adolescents. J Pediatr 154 (5): 708-14, 2009.
-
Shayota BJ, Pawar SC, Chamberlain RS: MeSS: A novel prognostic scale specific for pediatric well-differentiated thyroid cancer: a population-based, SEER outcomes study. Surgery 154 (3): 429-35, 2013.
-
Sassolas G, Hafdi-Nejjari Z, Casagranda L, et al.: Thyroid cancers in children, adolescents, and young adults with and without a history of childhood exposure to therapeutic radiation for other cancers. Thyroid 23 (7): 805-10, 2013.
-
Willgerodt H, Keller E, Bennek J, et al.: Diagnostic value of fine-needle aspiration biopsy of thyroid nodules in children and adolescents. J Pediatr Endocrinol Metab 19 (4): 507-15, 2006.
-
Stevens C, Lee JK, Sadatsafavi M, et al.: Pediatric thyroid fine-needle aspiration cytology: a meta-analysis. J Pediatr Surg 44 (11): 2184-91, 2009.
-
Bargren AE, Meyer-Rochow GY, Sywak MS, et al.: Diagnostic utility of fine-needle aspiration cytology in pediatric differentiated thyroid cancer. World J Surg 34 (6): 1254-60, 2010.
-
Redlich A, Boxberger N, Kurt Werner S, et al.: Sensitivity of fine-needle biopsy in detecting pediatric differentiated thyroid carcinoma. Pediatr Blood Cancer 59 (2): 233-7, 2012.
-
Waguespack SG, Francis G: Initial management and follow-up of differentiated thyroid cancer in children. J Natl Compr Canc Netw 8 (11): 1289-300, 2010.
-
Vassilopoulou-Sellin R, Goepfert H, Raney B, et al.: Differentiated thyroid cancer in children and adolescents: clinical outcome and mortality after long-term follow-up. Head Neck 20 (6): 549-55, 1998.
-
Wiersinga WM: Thyroid cancer in children and adolescents--consequences in later life. J Pediatr Endocrinol Metab 14 (Suppl 5): 1289-96; discussion 1297-8, 2001.
-
Jarzab B, Handkiewicz-Junak D, Wloch J: Juvenile differentiated thyroid carcinoma and the role of radioiodine in its treatment: a qualitative review. Endocr Relat Cancer 12 (4): 773-803, 2005.
-
Alessandri AJ, Goddard KJ, Blair GK, et al.: Age is the major determinant of recurrence in pediatric differentiated thyroid carcinoma. Med Pediatr Oncol 35 (1): 41-6, 2000.
-
Borson-Chazot F, Causeret S, Lifante JC, et al.: Predictive factors for recurrence from a series of 74 children and adolescents with differentiated thyroid cancer. World J Surg 28 (11): 1088-92, 2004.
-
Biko J, Reiners C, Kreissl MC, et al.: Favourable course of disease after incomplete remission on (131)I therapy in children with pulmonary metastases of papillary thyroid carcinoma: 10 years follow-up. Eur J Nucl Med Mol Imaging 38 (4): 651-5, 2011.
-
Patel A, Jhiang S, Dogra S, et al.: Differentiated thyroid carcinoma that express sodium-iodide symporter have a lower risk of recurrence for children and adolescents. Pediatr Res 52 (5): 737-44, 2002.
-
Powers PA, Dinauer CA, Tuttle RM, et al.: Treatment of recurrent papillary thyroid carcinoma in children and adolescents. J Pediatr Endocrinol Metab 16 (7): 1033-40, 2003.
-
Kloos RT, Ringel MD, Knopp MV, et al.: Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol 27 (10): 1675-84, 2009.
-
Waguespack SG, Sherman SI, Williams MD, et al.: The successful use of sorafenib to treat pediatric papillary thyroid carcinoma. Thyroid 19 (4): 407-12, 2009.
-
Brose MS, Nutting CM, Jarzab B, et al.: Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384 (9940): 319-28, 2014.
-
Iyer P, Mayer JL, Ewig JM: Response to sorafenib in a pediatric patient with papillary thyroid carcinoma with diffuse nodular pulmonary disease requiring mechanical ventilation. Thyroid 24 (1): 169-74, 2014.
-
Schlumberger M, Tahara M, Wirth LJ, et al.: Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 372 (7): 621-30, 2015.
-
Falchook GS, Long GV, Kurzrock R, et al.: Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379 (9829): 1893-901, 2012.
-
Hayes DN, Lucas AS, Tanvetyanon T, et al.: Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res 18 (7): 2056-65, 2012.
-
Waguespack SG, Rich TA, Perrier ND, et al.: Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol 7 (10): 596-607, 2011.
-
Krueger JE, Maitra A, Albores-Saavedra J: Inherited medullary microcarcinoma of the thyroid: a study of 11 cases. Am J Surg Pathol 24 (6): 853-8, 2000.
-
Brauckhoff M, Machens A, Lorenz K, et al.: Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg 259 (4): 800-6, 2014.
-
Raval MV, Sturgeon C, Bentrem DJ, et al.: Influence of lymph node metastases on survival in pediatric medullary thyroid cancer. J Pediatr Surg 45 (10): 1947-54, 2010.
-
Wells SA Jr, Robinson BG, Gagel RF, et al.: Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30 (2): 134-41, 2012.
-
Thornton K, Kim G, Maher VE, et al.: Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res 18 (14): 3722-30, 2012.
-
Fox E, Widemann BC, Chuk MK, et al.: Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 19 (15): 4239-48, 2013.
-
Kurzrock R, Sherman SI, Ball DW, et al.: Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 29 (19): 2660-6, 2011.
-
Das S, Das AK: A review of pediatric oral biopsies from a surgical pathology service in a dental school. Pediatr Dent 15 (3): 208-11, 1993 May-Jun.
-
Ulmansky M, Lustmann J, Balkin N: Tumors and tumor-like lesions of the oral cavity and related structures in Israeli children. Int J Oral Maxillofac Surg 28 (4): 291-4, 1999.
-
Tröbs RB, Mader E, Friedrich T, et al.: Oral tumors and tumor-like lesions in infants and children. Pediatr Surg Int 19 (9-10): 639-45, 2003.
-
Tanaka N, Murata A, Yamaguchi A, et al.: Clinical features and management of oral and maxillofacial tumors in children. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 88 (1): 11-5, 1999.
-
Young JL Jr, Miller RW: Incidence of malignant tumors in U. S. children. J Pediatr 86 (2): 254-8, 1975.
-
Berstein L, Gurney JG: Carcinomas and other malignant epithelial neoplasms. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, Chapter 11, pp 139-148. Also available online. Last accessed April 04, 2017.
-
Bleyer A: Cancer of the oral cavity and pharynx in young females: increasing incidence, role of human papilloma virus, and lack of survival improvement. Semin Oncol 36 (5): 451-9, 2009.
-
D'Souza G, Dempsey A: The role of HPV in head and neck cancer and review of the HPV vaccine. Prev Med 53 (Suppl 1): S5-S11, 2011.
-
Gillison ML, Broutian T, Pickard RK, et al.: Prevalence of oral HPV infection in the United States, 2009-2010. JAMA 307 (7): 693-703, 2012.
-
Simard EP, Ward EM, Siegel R, et al.: Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin 62 (2): 118-28, 2012 Mar-Apr.
-
Gillison ML, Chaturvedi AK, Lowy DR: HPV prophylactic vaccines and the potential prevention of noncervical cancers in both men and women. Cancer 113 (10 Suppl): 3036-46, 2008.
-
Morris LG, Ganly I: Outcomes of oral cavity squamous cell carcinoma in pediatric patients. Oral Oncol 46 (4): 292-6, 2010.
-
Perez DE, Pires FR, Alves Fde A, et al.: Juvenile intraoral mucoepidermoid carcinoma. J Oral Maxillofac Surg 66 (2): 308-11, 2008.
-
Oksüzoğlu B, Yalçin S: Squamous cell carcinoma of the tongue in a patient with Fanconi's anemia: a case report and review of the literature. Ann Hematol 81 (5): 294-8, 2002.
-
Reinhard H, Peters I, Gottschling S, et al.: Squamous cell carcinoma of the tongue in a 13-year-old girl with Fanconi anemia. J Pediatr Hematol Oncol 29 (7): 488-91, 2007.
-
Ragin CC, Modugno F, Gollin SM: The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res 86 (2): 104-14, 2007.
-
Fine JD, Johnson LB, Weiner M, et al.: Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol 60 (2): 203-11, 2009.
-
Kraemer KH, Lee MM, Scotto J: Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 123 (2): 241-50, 1987.
-
Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
-
Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, et al.: Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol 156 (5): 1015-9, 2007.
-
Alter BP, Giri N, Savage SA, et al.: Cancer in dyskeratosis congenita. Blood 113 (26): 6549-57, 2009.
-
Sturgis EM, Moore BA, Glisson BS, et al.: Neoadjuvant chemotherapy for squamous cell carcinoma of the oral tongue in young adults: a case series. Head Neck 27 (9): 748-56, 2005.
-
Woo VL, Kelsch RD, Su L, et al.: Gingival squamous cell carcinoma in adolescence. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 107 (1): 92-9, 2009.
-
Sultan I, Rodriguez-Galindo C, Al-Sharabati S, et al.: Salivary gland carcinomas in children and adolescents: a population-based study, with comparison to adult cases. Head Neck 33 (10): 1476-81, 2011.
-
Cesmebasi A, Gabriel A, Niku D, et al.: Pediatric head and neck tumors: an intra-demographic analysis using the SEER* database. Med Sci Monit 20: 2536-42, 2014.
-
Kaste SC, Hedlund G, Pratt CB: Malignant parotid tumors in patients previously treated for childhood cancer: clinical and imaging findings in eight cases. AJR Am J Roentgenol 162 (3): 655-9, 1994.
-
Whatley WS, Thompson JW, Rao B: Salivary gland tumors in survivors of childhood cancer. Otolaryngol Head Neck Surg 134 (3): 385-8, 2006.
-
Chowdhry AK, McHugh C, Fung C, et al.: Second primary head and neck cancer after Hodgkin lymphoma: a population-based study of 44,879 survivors of Hodgkin lymphoma. Cancer 121 (9): 1436-45, 2015.
-
Rutt AL, Hawkshaw MJ, Lurie D, et al.: Salivary gland cancer in patients younger than 30 years. Ear Nose Throat J 90 (4): 174-84, 2011.
-
Allan BJ, Tashiro J, Diaz S, et al.: Malignant tumors of the parotid gland in children: incidence and outcomes. J Craniofac Surg 24 (5): 1660-4, 2013.
-
Ethunandan M, Ethunandan A, Macpherson D, et al.: Parotid neoplasms in children: experience of diagnosis and management in a district general hospital. Int J Oral Maxillofac Surg 32 (4): 373-7, 2003.
-
da Cruz Perez DE, Pires FR, Alves FA, et al.: Salivary gland tumors in children and adolescents: a clinicopathologic and immunohistochemical study of fifty-three cases. Int J Pediatr Otorhinolaryngol 68 (7): 895-902, 2004.
-
Muenscher A, Diegel T, Jaehne M, et al.: Benign and malignant salivary gland diseases in children A retrospective study of 549 cases from the Salivary Gland Registry, Hamburg. Auris Nasus Larynx 36 (3): 326-31, 2009.
-
Fu H, Wang J, Wang L, et al.: Pleomorphic adenoma of the salivary glands in children and adolescents. J Pediatr Surg 47 (4): 715-9, 2012.
-
Galer C, Santillan AA, Chelius D, et al.: Minor salivary gland malignancies in the pediatric population. Head Neck 34 (11): 1648-51, 2012.
-
Thariat J, Vedrine PO, Temam S, et al.: The role of radiation therapy in pediatric mucoepidermoid carcinomas of the salivary glands. J Pediatr 162 (4): 839-43, 2013.
-
Chiaravalli S, Guzzo M, Bisogno G, et al.: Salivary gland carcinomas in children and adolescents: the Italian TREP project experience. Pediatr Blood Cancer 61 (11): 1961-8, 2014.
-
Laikui L, Hongwei L, Hongbing J, et al.: Epithelial salivary gland tumors of children and adolescents in west China population: a clinicopathologic study of 79 cases. J Oral Pathol Med 37 (4): 201-5, 2008.
-
Rahbar R, Grimmer JF, Vargas SO, et al.: Mucoepidermoid carcinoma of the parotid gland in children: A 10-year experience. Arch Otolaryngol Head Neck Surg 132 (4): 375-80, 2006.
-
Kupferman ME, de la Garza GO, Santillan AA, et al.: Outcomes of pediatric patients with malignancies of the major salivary glands. Ann Surg Oncol 17 (12): 3301-7, 2010.
-
Aro K, Leivo I, Mäkitie A: Management of salivary gland malignancies in the pediatric population. Curr Opin Otolaryngol Head Neck Surg 22 (2): 116-20, 2014.
-
Techavichit P, Hicks MJ, López-Terrada DH, et al.: Mucoepidermoid Carcinoma in Children: A Single Institutional Experience. Pediatr Blood Cancer 63 (1): 27-31, 2016.
-
Verma J, Teh BS, Paulino AC: Characteristics and outcome of radiation and chemotherapy-related mucoepidermoid carcinoma of the salivary glands. Pediatr Blood Cancer 57 (7): 1137-41, 2011.
-
Védrine PO, Coffinet L, Temam S, et al.: Mucoepidermoid carcinoma of salivary glands in the pediatric age group: 18 clinical cases, including 11 second malignant neoplasms. Head Neck 28 (9): 827-33, 2006.
-
Ryan JT, El-Naggar AK, Huh W, et al.: Primacy of surgery in the management of mucoepidermoid carcinoma in children. Head Neck 33 (12): 1769-73, 2011.
-
Grant SR, Grosshans DR, Bilton SD, et al.: Proton versus conventional radiotherapy for pediatric salivary gland tumors: Acute toxicity and dosimetric characteristics. Radiother Oncol 116 (2): 309-15, 2015.
-
Williams SB, Ellis GL, Warnock GR: Sialoblastoma: a clinicopathologic and immunohistochemical study of 7 cases. Ann Diagn Pathol 10 (6): 320-6, 2006.
-
Prigent M, Teissier N, Peuchmaur M, et al.: Sialoblastoma of salivary glands in children: chemotherapy should be discussed as an alternative to mutilating surgery. Int J Pediatr Otorhinolaryngol 74 (8): 942-5, 2010.
-
Scott JX, Krishnan S, Bourne AJ, et al.: Treatment of metastatic sialoblastoma with chemotherapy and surgery. Pediatr Blood Cancer 50 (1): 134-7, 2008.
-
Bitar MA, Moukarbel RV, Zalzal GH: Management of congenital subglottic hemangioma: trends and success over the past 17 years. Otolaryngol Head Neck Surg 132 (2): 226-31, 2005.
-
McGuirt WF Jr, Little JP: Laryngeal cancer in children and adolescents. Otolaryngol Clin North Am 30 (2): 207-14, 1997.
-
Bauman NM, Smith RJ: Recurrent respiratory papillomatosis. Pediatr Clin North Am 43 (6): 1385-401, 1996.
-
Pappo AS, Meza JL, Donaldson SS, et al.: Treatment of localized nonorbital, nonparameningeal head and neck rhabdomyosarcoma: lessons learned from intergroup rhabdomyosarcoma studies III and IV. J Clin Oncol 21 (4): 638-45, 2003.
-
Siddiqui F, Sarin R, Agarwal JP, et al.: Squamous carcinoma of the larynx and hypopharynx in children: a distinct clinical entity? Med Pediatr Oncol 40 (5): 322-4, 2003.
-
Kashima HK, Mounts P, Shah K: Recurrent respiratory papillomatosis. Obstet Gynecol Clin North Am 23 (3): 699-706, 1996.
-
Maloney EM, Unger ER, Tucker RA, et al.: Longitudinal measures of human papillomavirus 6 and 11 viral loads and antibody response in children with recurrent respiratory papillomatosis. Arch Otolaryngol Head Neck Surg 132 (7): 711-5, 2006.
-
Gélinas JF, Manoukian J, Côté A: Lung involvement in juvenile onset recurrent respiratory papillomatosis: a systematic review of the literature. Int J Pediatr Otorhinolaryngol 72 (4): 433-52, 2008.
-
Andrus JG, Shapshay SM: Contemporary management of laryngeal papilloma in adults and children. Otolaryngol Clin North Am 39 (1): 135-58, 2006.
-
Avidano MA, Singleton GT: Adjuvant drug strategies in the treatment of recurrent respiratory papillomatosis. Otolaryngol Head Neck Surg 112 (2): 197-202, 1995.
-
Derkay CS, Smith RJ, McClay J, et al.: HspE7 treatment of pediatric recurrent respiratory papillomatosis: final results of an open-label trial. Ann Otol Rhinol Laryngol 114 (9): 730-7, 2005.
-
Sidell DR, Nassar M, Cotton RT, et al.: High-dose sublesional bevacizumab (avastin) for pediatric recurrent respiratory papillomatosis. Ann Otol Rhinol Laryngol 123 (3): 214-21, 2014.
-
Chadha NK, James A: Adjuvant antiviral therapy for recurrent respiratory papillomatosis. Cochrane Database Syst Rev 12: CD005053, 2012.
-
French CA, Rahman S, Walsh EM, et al.: NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov 4 (8): 928-41, 2014.
-
French CA: NUT midline carcinoma. Cancer Genet Cytogenet 203 (1): 16-20, 2010.
-
French CA, Kutok JL, Faquin WC, et al.: Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol 22 (20): 4135-9, 2004.
-
Bauer DE, Mitchell CM, Strait KM, et al.: Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin Cancer Res 18 (20): 5773-9, 2012.
-
Schwartz BE, Hofer MD, Lemieux ME, et al.: Differentiation of NUT midline carcinoma by epigenomic reprogramming. Cancer Res 71 (7): 2686-96, 2011.
-
Maher OM, Christensen AM, Yedururi S, et al.: Histone deacetylase inhibitor for NUT midline carcinoma. Pediatr Blood Cancer 62 (4): 715-7, 2015.
Thoracic CancersThoracic cancers include the following: - Breast cancer.
- Lung cancer.
- Bronchial tumors.
- Pleuropulmonary blastoma.
- Esophageal tumors.
- Thymoma and thymic carcinoma.
- Cardiac tumors.
- Mesothelioma.
The prognosis, diagnosis, classification, and treatment of these thoracic cancers are discussed below. It must be emphasized that these cancers are seen very infrequently in patients younger than 15 years, and most of the evidence is derived from case series.[1] Breast Cancer Fibroadenoma The most frequent breast tumor seen in children is a fibroadenoma.[2,3] These tumors can be observed and many will regress without a need for surgical resection. However, rare malignant transformation leading to phyllodes tumors has been reported.[4] Sudden rapid enlargement of a suspected fibroadenoma is an indication for needle biopsy or excision. Treatment of Fibroadenoma Phyllodes tumors can be managed by wide local excision without mastectomy.[4] Breast Cancer Incidence and Outcome Breast cancer has been reported in both males and females younger than 21 years.[5,6,7,8,9,10] A review of the Surveillance, Epidemiology, and End Results (SEER) database of the National Cancer Institute shows that 75 cases of malignant breast tumors in females aged 19 years or younger were identified from 1973 to 2004.[11] Fifteen percent of these patients had in situ disease, 85% had invasive disease, 55% of the tumors were carcinomas, and 45% of the tumors were sarcomas-most of which were phyllodes tumors. Only three patients in the carcinoma group presented with metastatic disease, while 11 patients (27%) had regionally advanced disease. All patients with sarcomas presented with localized disease. Of the carcinoma patients, 85% underwent surgical resection, and 10% received adjuvant radiation therapy. Of the sarcoma patients, 97% had surgical resection, and 9% received radiation. The 5- and 10-year survival rates for patients with sarcomatous tumors were both 90%; for patients with carcinomas, the 5-year survival rate was 63% and the 10-year survival rate was 54%. Breast tumors may also occur as metastatic deposits from leukemia, rhabdomyosarcoma, other sarcomas, or lymphoma (particularly in patients who are infected with the human immunodeficiency virus). Risk Factors Risk factors for breast cancer in adolescents and young adults include the following: - Previous malignancy. A retrospective review of the American College of Surgeons National Cancer Database from 1998 to 2010 identified 106,771 patients aged 15 to 39 years with breast cancer.[12] Of these patients, 6,241 (5.8%) had experienced a previous histologically distinct malignancy. Patients with breast cancer as a subsequent neoplasm had a significantly decreased 3-year overall survival (OS) (79% vs. 88.5%, P <.001), with subsequent neoplasm status identified as an independent risk factor for increased mortality (hazard ratio, 1.58; 95% confidence interval, 1.41-1.77).
- Chest irradiation. There is an increased lifetime risk of breast cancer in female survivors of Hodgkin lymphoma who were treated with radiation to the chest area; however, breast cancer is also seen in patients who were treated for any cancer that was treated with chest irradiation.[9,13,14,15,16][Level of evidence: 1A] Carcinomas are more frequent than sarcomas.
Mammograms with adjunctive breast magnetic resonance imaging (MRI) start at age 25 years or 10 years postexposure to radiation therapy (whichever came last). (Refer to the PDQ summary on the Late Effects of Treatment for Childhood Cancer for more information about secondary breast cancers.)
Treatment of Breast Cancer in Adolescents and Young Adults (AYA) Breast cancer is the most frequently diagnosed cancer among AYA women aged 15 to 39 years, accounting for about 14% of all AYA cancer diagnoses.[17] Breast cancer in this age group has a more aggressive course and worse outcome than in older women. Expression of hormone receptors for estrogen, progesterone, and human epidermal growth factor 2 (HER2) on breast cancer in the AYA group is also different from that in older women and correlates with a worse prognosis.[12,18] Treatment of the AYA group is similar to that of older women. However, unique aspects of management must include attention to genetic implications (i.e., familial breast cancer syndromes) and fertility.[19,20] (Refer to the PDQ summary on adult Breast Cancer Treatment or the PDQ summary on Genetics of Breast and Gynecologic Cancers for more information.) Lung Cancer Primary lung tumors are rare in children and histologically quite diverse.[1] When epithelial cancers of the lung occur, they tend to be of advanced stage, with prognosis dependent on both histology and stage.[21] Most primary lung tumors are malignant. In a review of 383 primary pulmonary neoplasms in children, 76% were malignant and 24% were benign.[22] A review of primary malignant epithelial lung tumors using the National Cancer Data Base found that the most common primary malignant pediatric lung neoplasms were carcinoid tumors (63%) followed by mucoepidermoid carcinoma of the lung (18%).[23] Most pulmonary malignant neoplasms in children are due to metastatic disease, with an approximate ratio of primary malignant tumors to metastatic disease of 1:5.[24] The most common malignant primary tumors of the lung are bronchial tumors and pleuropulmonary blastoma. Bronchial Tumors Histology Bronchial tumors are a heterogeneous group of primary endobronchial lesions, and although adenoma implies a benign process, all varieties of bronchial tumors on occasion display malignant behavior. The following three histologic types have been identified:[25,26,27,28,29,30] - Carcinoid tumor (neuroendocrine tumor of the bronchus). Carcinoid tumors account for 80% to 85% of all bronchial tumors in children.[25,26,27,28,29] It is the most common bronchial tumor.
- Mucoepidermoid carcinoma.
- Adenoid cystic carcinoma. It is the least common bronchial tumor.
Prognosis Bronchial tumors of all histologic types are associated with an excellent prognosis after surgical resection in children, even in the presence of local invasion.[31,32]; [33][Level of evidence: 2A] Clinical Presentation and Diagnostic Evaluation The presenting symptoms of a bronchial tumor are usually caused by an incomplete bronchial obstruction and include the following: - Cough.
- Recurrent pneumonitis.
- Hemoptysis.
Because of difficulties in diagnosis, symptoms are frequently present for months, and, occasionally, children with wheezing have been treated for asthma, with delays in diagnosis for as long as 4 to 5 years.[34] Metastatic lesions are reported in approximately 6% of carcinoid tumors, and recurrences are reported in 2% of cases. Atypical carcinoid tumors are rare but more aggressive, with 50% of patients presenting with metastatic disease at diagnosis.[21,35] There is a single report of a child with a carcinoid tumor and metastatic disease who developed the classic carcinoid syndrome.[36] Octreotide nuclear scans may demonstrate uptake of radioactivity by the tumor or lymph nodes, suggesting metastatic spread. The management of bronchial tumors is somewhat controversial because bronchial tumors are usually visible endoscopically. Biopsy of these lesions may be hazardous because of the risk of hemorrhage. New endoscopic techniques have allowed biopsy to be performed safely;[30,37] however, endoscopic resection is not recommended except in highly selected cases.[37,38] Bronchography or computed tomography scan may be helpful to determine the degree of bronchiectasis distal to the obstruction since the degree of pulmonary destruction may influence surgical therapy.[39] Treatment Conservative pulmonary resection, including sleeve segmental resection, when feasible, with the removal of the involved lymphatics, is the treatment of choice.[40,41]; [33][Level of evidence: 2A] Adenoid cystic carcinomas (cylindroma) have a tendency to spread submucosally, and late local recurrence or dissemination has been reported. In addition to en bloc resection with hilar lymphadenectomy, a frozen section examination of the bronchial margins is performed in children with this lesion. Neither chemotherapy nor radiation therapy is indicated for bronchial tumors, unless evidence of metastasis is documented. (Refer to the Neuroendocrine Tumors (Carcinoid Tumors) section of this summary for information about neuroendocrine carcinoid tumors.) Pleuropulmonary Blastoma Types of Pleuropulmonary Blastoma Pleuropulmonary blastoma is a rare and highly aggressive pulmonary malignancy that can present as a pulmonary or pleural mass. The International Pleuropulmonary Blastoma Registry is a valuable resource for information on this rare malignancy.[42] The following three subtypes of pleuropulmonary blastoma have been identified: - Type I: A purely lung cystic neoplasm with subtle malignant changes that typically occurs in the first 2 years of life and has a good prognosis. Transition from Type I to Type III occurs;[43,44] however, a significant proportion of type I lesions may not progress to types II and III.[44]
Histologically, these tumors appear as a multilocular cyst with variable numbers of primitive mesenchymal cells beneath a benign epithelial surface, with skeletal differentiation in one-half of the cases.[44] This form of disease can be clinically and pathologically deceptive because of its resemblance to some developmental lung cysts. - Type Ir: A purely cystic tumor that lacks a primitive cell component. The r designation signifies regression or nonprogression. Type Ir was originally recognized in older siblings of pleuropulmonary blastoma patients, but can be seen in very young children. A lung cyst in an older individual with a DICER1 mutation or in a relative of a pleuropulmonary blastoma patient is most likely to be type Ir.[45]
- Type II: Type II exhibits both cystic and solid components. The solid areas have mixed blastomatous and sarcomatous features; most of the cases exhibit rhabdomyoblasts, and nodules with cartilaginous differentiation are common.[46] Cerebral metastasis may occur in 11% of patients.[47]
- Type III: A purely solid neoplasm, with the blastomatous and sarcomatous elements described above.[48,49] Cerebral metastasis occurs in up to 50% of patients with Type III tumors.[47]
The Pleuropulmonary Blastoma Registry reported on 350 centrally reviewed and confirmed cases of pleuropulmonary blastoma over a 50 year period.[45] Table 4. Relative Proportions and Features of Pleuropulmonary Blastomaa | Type I | Type Ir | Type II | Type II/III or III |
|---|
| a Adapted from Messinger et al.[45] | | Relative proportion of pleuropulmonary blastoma cases | 33% | 35% | 32% | | Median age at diagnosis (months) | 8 | 47 | 35 | 41 | | 5-year overall survival | 89% | 100% | 71% | 53% | Prognostic Factors Prognostic factors for pleuropulmonary blastoma include the following:[45] - Type of pleuropulmonary blastoma. The pleuropulmonary blastoma type was the strongest predictor of outcome. (Refer to Table 4.)
- Presence of metastatic disease. The presence of metastatic disease at diagnosis was also an independent unfavorable prognostic factor. There is an increased risk of developing brain metastases, with a 5-year cumulative probability of 11% for Type II disease and 54% for Type III disease.[47]
- Complete surgical resection.[50]
Of the 97 patients who were tested, 66% had a heterozygous germline DICER1 mutation, confirming that this is a familial cancer syndrome.[45] In this subset, DICER1 mutation status was not related to outcome. Risk Factors Approximately one-third of families of children with pleuropulmonary blastoma manifest a number of dysplastic and/or neoplastic conditions comprising the pleuropulmonary blastoma family tumor and dysplasia syndrome. Germline mutations in the DICER1 gene are considered the major genetic determinant of the complex.[51,52,53] Importantly, while DICER1 mutations cause a wide range of phenotypes, pleuropulmonary blastoma does not occur in all families with DICER1 mutations; therefore, the term DICER1 syndrome is generally used for these families. Also, most mutation carriers are unaffected, indicating that tumor risk is modest.[52] Conversely, approximately 40% of patients with pleuropulmonary blastoma tumors do not have DICER1 germline mutations.[45] The most relevant association is with cystic nephroma; up to 10% of pleuropulmonary blastoma cases have been reported to develop cystic nephroma or Wilms tumor, malignancies that are also more prevalent among family members.[54] Germline DICER1 mutations have also been associated with ovarian sex cord-stromal tumors (especially Sertoli-Leydig cell tumor), multinodular goiter, uterine cervix embryonal rhabdomyosarcoma, cervical primitive neuroectodermal tumor, Wilms tumor, pulmonary sequestration, juvenile intestinal polyps, ciliary body medulloepithelioma, medulloblastoma, and seminoma.[46,53,54,55,56,57,58,59] DICER1 mutations appear to have a low penetrance, with pleuropulmonary blastoma, cystic nephroma, and multinodular goiter being the most frequently reported manifestations; not all families include pleuropulmonary blastoma, and most mutation carriers do not develop tumors. Most associated conditions occur in children younger than 10 years, although ovarian tumors and multinodular goiters are described in children and adults aged up to 30 years.[53] Clinical Presentation Presenting symptoms are not specific, and commonly include the following: - Respiratory distress.
- Fever.
- Chest pain.
Up to 50% of patients with type I disease have multiple lesions, and in 33% of the cases lesions are bilateral.[44] The tumor is usually located in the lung periphery, but it may be extrapulmonary with involvement of the heart/great vessels, mediastinum, diaphragm, and/or pleura.[50,60] The International Pleuropulmonary Blastoma Registry identified 11 cases of Type II and Type III pleuropulmonary blastoma with tumor extension into the thoracic great vessels or the heart. Radiographic evaluation of the central circulation is performed in children with suspected or diagnosed pleuropulmonary blastoma to identify potentially fatal embolic complications.[61] Treatment There are no standard treatment options. Current treatment regimens for these rare tumors have been informed by consensus opinion. A complete surgical resection is the most important prognostic factor;[50] however, surgery alone results in high relapse rates.[44,49] Data from the International Pleuropulmonary Blastoma Registry and from the European Cooperative Study Group in Pediatric Rare Tumors (EXPeRT) suggest that adjuvant chemotherapy may reduce the risk of recurrence.[48]; [60][Level of evidence: 3iiiA] Responses to chemotherapy have been reported with agents similar to those used for the treatment of rhabdomyosarcoma.[45,48,62,63] High-dose chemotherapy with stem cell rescue has been used without success.[64] Some general treatment considerations from the Pleuropulmonary Blastoma Registry include the following:[42] - Type I and Type Ir: Surgery alone for select cases, particularly for Type Ir. However, adjuvant chemotherapy may decrease the risk of recurrence but does not affect survival.[42,45,48] Evidence suggests a close histologic relationship between a Type 4 cystic adenomatoid malformation and a Type I pleuropulmonary blastoma.[65,66] Complete surgical lobectomy is adequate treatment for these patients, but close observation is recommended.
- Type II and Type III: Surgery and chemotherapy, either preoperative or in the adjuvant setting.[45,62] A rhabdomyosarcoma regimen with complete surgical resection and chemotherapy with an anthracycline-containing regimen have been associated with better outcomes.[60]
Radiation therapy may be used in patients with type III pleuropulmonary blastoma, although this has no impact on survival.[45] Treatment Options Under Clinical Evaluation The following is an example of a national and/or institutional clinical trial that is currently being conducted. Information about ongoing clinical trials is available from the NCI website. - ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of the IMGN901 antibody-drug conjugate, which links a potent antimitotic drug to antibodies that target CD56.
Esophageal Tumors Incidence and Histology Esophageal cancer is rare in the pediatric age group, although it is relatively common in older adults.[67,68] Most of these tumors are squamous cell carcinomas, although sarcomas can also arise in the esophagus. The most common benign tumor is leiomyoma. Clinical Presentation and Diagnostic Evaluation Symptoms are related to difficulty in swallowing and associated weight loss. Diagnosis is made by histologic examination of biopsy tissue. Treatment Treatment options for esophageal carcinoma include the following: - External-beam intracavitary radiation therapy.
- Chemotherapy (agents commonly used to treat carcinomas such as platinum derivatives, paclitaxel, and etoposide).
- Surgery.
Prognosis is generally poor for this cancer, which rarely can be completely resected. (Refer to the PDQ summary on adult Esophageal Cancer Treatment for more information.) Thymoma and Thymic Carcinoma A cancer of the thymus is not considered a thymoma or a thymic carcinoma unless there are neoplastic changes of the epithelial cells that cover the organ.[69,70,71] The term thymoma is customarily used to describe neoplasms that show no overt atypia of the epithelial component. Thymic carcinomas have a higher incidence of capsular invasion and metastases. A thymic epithelial tumor that exhibits clear-cut cytologic atypia and histologic features no longer specific to the thymus is known as thymic carcinoma or type C thymoma. Other tumors that involve the thymus gland include lymphomas, germ cell tumors, carcinomas, carcinoids, and thymomas. Hodgkin lymphoma and non-Hodgkin lymphoma may also involve the thymus and must be differentiated from true thymomas and thymic carcinomas. Thymoma Incidence and outcome Thymomas are very rare in children.[72,73,74] In the Tumori Rari in Età Pediatrica registry, only eight cases were identified over a 9-year period.[72] Several studies have reported on outcomes associated with thymomas: - A review of the SEER registry from 1973 to 2008 identified 73 cases of malignant anterior mediastinal tumors in patients younger than 20 years.[73] Of these cases, 32% were thymomas, 29% were non-Hodgkin lymphoma, and 22% were Hodgkin lymphoma. Patients with thymomas had a worse survival at 10 years than did patients with lymphomas. Patients with thymoma who were treated in an earlier era from 1973 to 1989 had a 10-year survival rate of 18%; patients who were treated between 1991 and 2008 had a 75% survival rate. Presence of metastatic disease and treatment without surgery were associated with a worse outcome.
- A review of 48 published cases of thymoma in patients younger than 18 years, excluding thymic carcinoma, found an association between stage of disease and survival; it also suggested guidelines for treatment. The overall 2-year survival in this series was 71%.[74]
- The European Cooperative Study Group for Pediatric Rare Tumors identified 16 children with thymoma between 2000 and 2012.[75] Complete resection was achieved in 11 of 16 patients with thymoma. Fourteen of the 16 patients with thymoma were alive and well at a median of 5 years from diagnosis.
Risk factors Various diseases and syndromes are associated with thymoma, including the following:[76,77,78] - Myasthenia gravis.
- Polymyositis.
- Systemic lupus erythematosus.
- Rheumatoid arthritis.
- Thyroiditis.
- Isaac syndrome.
- Neuromyotonia (continuous muscle stiffness resulting from persistent muscle activity as a consequence of antibodies against voltage-gated potassium channels).
- Pure red-cell aplasia.
- Endocrine (hormonal) disorders such as hyperthyroidism, Addison disease, and panhypopituitarism.
Clinical presentation These neoplasms are usually located in the anterior mediastinum and discovered during a routine chest x-ray. Symptoms may include the following:[74] - Cough.
- Difficulty with swallowing.
- Tightness of the chest.
- Chest pain.
- Shortness of breath
Nonspecific symptoms may also occur. These tumors generally are slow growing but are potentially invasive, with metastases to distant organs or lymph nodes. Staging is related to invasiveness. Most children present with low-stage disease.[74] Treatment Treatment options for thymoma include the following: - Surgery. Surgery is performed with the goal of a complete resection and is the mainstay of therapy.[79]
- Radiation therapy. Radiation therapy is used in patients with invasive thymoma.[78]
- Chemotherapy. Chemotherapy is usually reserved for patients with advanced-stage disease who have not responded to radiation therapy or corticosteroids. Agents that have been effective include doxorubicin, cyclophosphamide, etoposide, cisplatin, ifosfamide, and vincristine.[72,78,80,81] Responses to regimens containing combinations of some of these agents have ranged from 26% to 100%, and survival rates have been as high as 50%.[81,82]
Thymic carcinoma The European Cooperative Study Group for Pediatric Rare Tumors identified 20 patients with thymic carcinoma between 2000 and 2012.[75] Complete resection was achieved in 1 of 20 patients with thymic carcinoma. A variety of chemotherapy regimens and radiation therapy were used for the patients with thymic carcinoma. Five-year OS for patients with thymic carcinoma was 21.0% ± 10.0%. Treatment Treatment options for thymic carcinoma include the following: - Surgery. Complete resection of thymic carcinoma is almost never possible.[75]
- Radiation therapy. Radiation therapy is used in patients with thymic carcinoma.[78]
- Chemotherapy (as described for thymoma). Response rates are lower for patients with thymic carcinoma, but 2-year survival rates have been reported to be as high as 50%.[71,83,84]
- Sunitinib. Sunitinib has yielded clinical responses in four adult patients with thymic carcinoma.[85]
(Refer to the PDQ summary on adult Thymoma and Thymic Carcinoma Treatment for more information on the treatment of thymoma and thymic carcinoma.) Cardiac Tumors Histology Cardiac tumors are rare, with an autopsy frequency of 0.001% to 0.30%;[86] in one report, the percentage of cardiac surgeries performed as a result of cardiac tumors was 0.093%.[87] The most common primary tumors of the heart are benign and include the following:[88,89,90] - Rhabdomyoma.
- Myxoma.
- Teratoma.
- Fibroma.
Other benign tumors include histiocytoid cardiomyopathy tumors, hemangiomas, and neurofibromas (i.e., tumors of the nerves that innervate the muscles).[88,91,92,93,94] Myxomas are the most common noncutaneous finding in Carney complex, a rare syndrome characterized by lentigines, cardiac myxomas or other myxoid fibromas, and endocrine abnormalities.[95,96,97] A mutation of the PRKAR1A gene is noted in more than 90% of the cases of Carney complex.[95,98] Primary malignant pediatric heart tumors are rare but may include the following:[88,99,100] - Malignant teratoma.
- Lymphoma.
- Various sarcomas, such as rhabdomyosarcoma, angiosarcoma, chondrosarcoma, and infantile fibrosarcoma. Rarely, synovial sarcoma may arise in the heart or pericardium.
Secondary tumors of the heart include metastatic spread of rhabdomyosarcoma, other sarcomas, melanoma, leukemia, thymoma, and carcinomas of various sites.[86,88] Risk Factors The distribution of cardiac tumors in the fetal and neonatal period is different from that in older patients, with two-thirds of teratomas occurring during this period of life.[91] Multiple cardiac tumors noted in the fetal or neonatal period are highly associated with a diagnosis of tuberous sclerosis.[91,101] A retrospective review of 94 patients with cardiac tumors detected by prenatal or neonatal echocardiography showed that 68% of the patients exhibited features of tuberous sclerosis.[102] In another study, 79% of patients (15 of 19) with rhabdomyomas discovered prenatally had tuberous sclerosis, while 96% of those diagnosed postnatally had tuberous sclerosis. Most rhabdomyomas, whether diagnosed prenatally or postnatally, will spontaneously regress.[103] Clinical Presentation and Diagnostic Evaluation Patients may be asymptomatic and present with sudden death,[104][Level of evidence: 3iiiA] but about two-thirds of patients have symptoms that may include the following: - Abnormalities of heart rhythm.
- Enlargement of the heart.
- Fluid in the pericardial sac.
- Congestive heart failure.
- Syncope.
- Stroke.
- Respiratory distress.[90]
The utilization of new cardiac MRI techniques can identify the likely tumor type in most children.[105] However, histologic diagnosis remains the standard for diagnosing cardiac tumors. Treatment Successful treatment may require surgery, debulking for progressive symptoms, cardiac transplantation, and chemotherapy that is appropriate for the type of cancer that is present:[106,107,108]; [109][Level of evidence: 3iiA] - Although some lesions such as rhabdomyomas can regress spontaneously, some practitioners recommend prophylactic resection to prevent mass-related complications.[87,90,101]; [110][Level of evidence: 3iiDiii] Treatment with the mammalian target of rapamycin (mTOR) inhibitor everolimus has been reported to be associated with a decrease in the size of rhabdomyomas in patients with tuberous sclerosis.[101,111]
- Cardiac sarcomas have a poor outcome and can be treated with multimodal therapy; the use of preoperative chemotherapy may be of value in reducing tumor volume before surgery.
- Complete surgical excision of other lesions offers the best chance for cure, with postoperative complications seen in about one-third of patients and postoperative mortality rates in less than 10% of patients.[87,90]
In one series, 95% of patients were free from cardiac tumor recurrence at 10 years.[90] Mesothelioma Incidence, Risk Factors, and Clinical Presentation Mesothelioma is extremely rare in childhood, with only 2% to 5% of patients presenting during the first two decades of life.[112] Fewer than 300 cases in children have been reported.[113] Mesothelioma may develop after successful treatment of an earlier cancer, especially after treatment with radiation.[114,115] In adults, these tumors have been associated with exposure to asbestos, which was used as building insulation.[116] The amount of exposure required to develop cancer is unknown, and there is no information about the risk for children exposed to asbestos. This tumor can involve the membranous coverings of the lung, the heart, or the abdominal organs.[117,118,119] These tumors can spread over the surface of organs, without invading far into the underlying tissue, and may spread to regional or distant lymph nodes. Prognosis Benign and malignant mesotheliomas cannot be differentiated using histologic criteria. A poor prognosis is associated with lesions that are diffuse and invasive and with those that recur. In general, the course of the disease is slow, and long-term survival is common. Diagnostic Evaluation Diagnostic thoracoscopy should be considered in suspicious cases to confirm diagnosis.[112] Treatment Radical surgical resection has been attempted with mixed results.[120] In adults, a multimodal therapy including extrapleural pneumonectomy and radiation therapy after combination chemotherapy with pemetrexed-cisplatin may achieve durable responses.[121][Level of evidence: 2A] However, this approach remains highly controversial.[122] In children, treatment with various chemotherapeutic agents used for carcinomas or sarcomas may result in partial responses.[119,123,124,125] Pain is an infrequent symptom; however, if pain occurs, radiation therapy may be used for palliation. Papillary serous carcinoma of the peritoneum may be mistaken for mesothelioma.[126] This tumor generally involves all surfaces lining the abdominal organs, including the surfaces of the ovary. Treatment includes surgical resection whenever possible and use of chemotherapy with agents such as cisplatin, carboplatin, and paclitaxel. (Refer to the PDQ summary on adult Malignant Mesothelioma Treatment for more information.) References:
-
Yu DC, Grabowski MJ, Kozakewich HP, et al.: Primary lung tumors in children and adolescents: a 90-year experience. J Pediatr Surg 45 (6): 1090-5, 2010.
-
Chung EM, Cube R, Hall GJ, et al.: From the archives of the AFIP: breast masses in children and adolescents: radiologic-pathologic correlation. Radiographics 29 (3): 907-31, 2009 May-Jun.
-
Jayasinghe Y, Simmons PS: Fibroadenomas in adolescence. Curr Opin Obstet Gynecol 21 (5): 402-6, 2009.
-
Valdes EK, Boolbol SK, Cohen JM, et al.: Malignant transformation of a breast fibroadenoma to cystosarcoma phyllodes: case report and review of the literature. Am Surg 71 (4): 348-53, 2005.
-
Serour F, Gilad A, Kopolovic J, et al.: Secretory breast cancer in childhood and adolescence: report of a case and review of the literature. Med Pediatr Oncol 20 (4): 341-4, 1992.
-
Drukker BH: Breast disease: a primer on diagnosis and management. Int J Fertil Womens Med 42 (5): 278-87, 1997 Sep-Oct.
-
Rogers DA, Lobe TE, Rao BN, et al.: Breast malignancy in children. J Pediatr Surg 29 (1): 48-51, 1994.
-
Rivera-Hueto F, Hevia-Vázquez A, Utrilla-Alcolea JC, et al.: Long-term prognosis of teenagers with breast cancer. Int J Surg Pathol 10 (4): 273-9, 2002.
-
Kaste SC, Hudson MM, Jones DJ, et al.: Breast masses in women treated for childhood cancer: incidence and screening guidelines. Cancer 82 (4): 784-92, 1998.
-
Costa NM, Rodrigues H, Pereira H, et al.: Secretory breast carcinoma--case report and review of the medical literature. Breast 13 (4): 353-5, 2004.
-
Gutierrez JC, Housri N, Koniaris LG, et al.: Malignant breast cancer in children: a review of 75 patients. J Surg Res 147 (2): 182-8, 2008.
-
Sadler C, Goldfarb M: Comparison of primary and secondary breast cancers in adolescents and young adults. Cancer 121 (8): 1295-302, 2015.
-
Metayer C, Lynch CF, Clarke EA, et al.: Second cancers among long-term survivors of Hodgkin's disease diagnosed in childhood and adolescence. J Clin Oncol 18 (12): 2435-43, 2000.
-
Swerdlow AJ, Barber JA, Hudson GV, et al.: Risk of second malignancy after Hodgkin's disease in a collaborative British cohort: the relation to age at treatment. J Clin Oncol 18 (3): 498-509, 2000.
-
van Leeuwen FE, Klokman WJ, Veer MB, et al.: Long-term risk of second malignancy in survivors of Hodgkin's disease treated during adolescence or young adulthood. J Clin Oncol 18 (3): 487-97, 2000.
-
Henderson TO, Amsterdam A, Bhatia S, et al.: Systematic review: surveillance for breast cancer in women treated with chest radiation for childhood, adolescent, or young adult cancer. Ann Intern Med 152 (7): 444-55; W144-54, 2010.
-
Keegan TH, DeRouen MC, Press DJ, et al.: Occurrence of breast cancer subtypes in adolescent and young adult women. Breast Cancer Res 14 (2): R55, 2012.
-
Anders CK, Hsu DS, Broadwater G, et al.: Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J Clin Oncol 26 (20): 3324-30, 2008.
-
Gabriel CA, Domchek SM: Breast cancer in young women. Breast Cancer Res 12 (5): 212, 2010.
-
Tichy JR, Lim E, Anders CK: Breast cancer in adolescents and young adults: a review with a focus on biology. J Natl Compr Canc Netw 11 (9): 1060-9, 2013.
-
Lal DR, Clark I, Shalkow J, et al.: Primary epithelial lung malignancies in the pediatric population. Pediatr Blood Cancer 45 (5): 683-6, 2005.
-
Hancock BJ, Di Lorenzo M, Youssef S, et al.: Childhood primary pulmonary neoplasms. J Pediatr Surg 28 (9): 1133-6, 1993.
-
Rojas Y, Shi YX, Zhang W, et al.: Primary malignant pulmonary tumors in children: a review of the national cancer data base. J Pediatr Surg 50 (6): 1004-8, 2015.
-
Weldon CB, Shamberger RC: Pediatric pulmonary tumors: primary and metastatic. Semin Pediatr Surg 17 (1): 17-29, 2008.
-
Vadasz P, Palffy G, Egervary M, et al.: Diagnosis and treatment of bronchial carcinoid tumors: clinical and pathological review of 120 operated patients. Eur J Cardiothorac Surg 7 (1): 8-11, 1993.
-
Kulke MH, Mayer RJ: Carcinoid tumors. N Engl J Med 340 (11): 858-68, 1999.
-
Oliaro A, Filosso PL, Donati G, et al.: Atypical bronchial carcinoids. Review of 46 patients. J Cardiovasc Surg (Torino) 41 (1): 131-5, 2000.
-
Moraes TJ, Langer JC, Forte V, et al.: Pediatric pulmonary carcinoid: a case report and review of the literature. Pediatr Pulmonol 35 (4): 318-22, 2003.
-
Al-Qahtani AR, Di Lorenzo M, Yazbeck S: Endobronchial tumors in children: Institutional experience and literature review. J Pediatr Surg 38 (5): 733-6, 2003.
-
Roby BB, Drehner D, Sidman JD: Pediatric tracheal and endobronchial tumors: an institutional experience. Arch Otolaryngol Head Neck Surg 137 (9): 925-9, 2011.
-
Soga J, Yakuwa Y: Bronchopulmonary carcinoids: An analysis of 1,875 reported cases with special reference to a comparison between typical carcinoids and atypical varieties. Ann Thorac Cardiovasc Surg 5 (4): 211-9, 1999.
-
Fauroux B, Aynie V, Larroquet M, et al.: Carcinoid and mucoepidermoid bronchial tumours in children. Eur J Pediatr 164 (12): 748-52, 2005.
-
Redlich A, Wechsung K, Boxberger N, et al.: Extra-appendiceal neuroendocrine neoplasms in children - data from the GPOH-MET 97 Study. Klin Padiatr 225 (6): 315-9, 2013.
-
Abuzetun JY, Hazin R, Suker M, et al.: Primary squamous cell carcinoma of the lung with bony metastasis in a 13-year-old boy: case report and review of literature. J Pediatr Hematol Oncol 30 (8): 635-7, 2008.
-
Rizzardi G, Marulli G, Calabrese F, et al.: Bronchial carcinoid tumours in children: surgical treatment and outcome in a single institution. Eur J Pediatr Surg 19 (4): 228-31, 2009.
-
Lack EE, Harris GB, Eraklis AJ, et al.: Primary bronchial tumors in childhood. A clinicopathologic study of six cases. Cancer 51 (3): 492-7, 1983.
-
Malkan AD, Sandoval JA: Controversial tumors in pediatric surgical oncology. Curr Probl Surg 51 (12): 478-520, 2014.
-
Luckraz H, Amer K, Thomas L, et al.: Long-term outcome of bronchoscopically resected endobronchial typical carcinoid tumors. J Thorac Cardiovasc Surg 132 (1): 113-5, 2006.
-
Ahel V, Zubovic I, Rozmanic V: Bronchial adenoid cystic carcinoma with saccular bronchiectasis as a cause of recurrent pneumonia in children. Pediatr Pulmonol 12 (4): 260-2, 1992.
-
Gaissert HA, Mathisen DJ, Grillo HC, et al.: Tracheobronchial sleeve resection in children and adolescents. J Pediatr Surg 29 (2): 192-7; discussion 197-8, 1994.
-
Jalal A, Jeyasingham K: Bronchoplasty for malignant and benign conditions: a retrospective study of 44 cases. Eur J Cardiothorac Surg 17 (4): 370-6, 2000.
-
Pleuropulmonary Blastoma Registry. St. Paul, Minn: Children's Hospitals and Clinics of St. Paul. Available online. Last accessed April 04, 2017.
-
Shivastava R, Saha A, Mehera B, et al.: Pleuropulmonary blastoma: transition from type I (cystic) to type III (solid). Singapore Med J 48 (7): e190-2, 2007.
-
Hill DA, Jarzembowski JA, Priest JR, et al.: Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am J Surg Pathol 32 (2): 282-95, 2008.
-
Messinger YH, Stewart DR, Priest JR, et al.: Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer 121 (2): 276-85, 2015.
-
Priest JR, McDermott MB, Bhatia S, et al.: Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer 80 (1): 147-61, 1997.
-
Priest JR, Magnuson J, Williams GM, et al.: Cerebral metastasis and other central nervous system complications of pleuropulmonary blastoma. Pediatr Blood Cancer 49 (3): 266-73, 2007.
-
Priest JR, Hill DA, Williams GM, et al.: Type I pleuropulmonary blastoma: a report from the International Pleuropulmonary Blastoma Registry. J Clin Oncol 24 (27): 4492-8, 2006.
-
Miniati DN, Chintagumpala M, Langston C, et al.: Prenatal presentation and outcome of children with pleuropulmonary blastoma. J Pediatr Surg 41 (1): 66-71, 2006.
-
Indolfi P, Bisogno G, Casale F, et al.: Prognostic factors in pleuro-pulmonary blastoma. Pediatr Blood Cancer 48 (3): 318-23, 2007.
-
Hill DA, Ivanovich J, Priest JR, et al.: DICER1 mutations in familial pleuropulmonary blastoma. Science 325 (5943): 965, 2009.
-
Slade I, Bacchelli C, Davies H, et al.: DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 48 (4): 273-8, 2011.
-
Foulkes WD, Bahubeshi A, Hamel N, et al.: Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32 (12): 1381-4, 2011.
-
Boman F, Hill DA, Williams GM, et al.: Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr 149 (6): 850-854, 2006.
-
Bouron-Dal Soglio D, Harvey I, Yazbeck S, et al.: An association of pleuropulmonary blastoma and cystic nephroma: possible genetic association. Pediatr Dev Pathol 9 (1): 61-4, 2006 Jan-Feb.
-
Priest JR, Williams GM, Manera R, et al.: Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma--a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol 95 (7): 1001-5, 2011.
-
Schultz KA, Pacheco MC, Yang J, et al.: Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 122 (2): 246-50, 2011.
-
Doros LA, Rossi CT, Yang J, et al.: DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 27 (9): 1267-80, 2014.
-
Doros L, Yang J, Dehner L, et al.: DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 59 (3): 558-60, 2012.
-
Bisogno G, Brennan B, Orbach D, et al.: Treatment and prognostic factors in pleuropulmonary blastoma: an EXPeRT report. Eur J Cancer 50 (1): 178-84, 2014.
-
Priest JR, Andic D, Arbuckle S, et al.: Great vessel/cardiac extension and tumor embolism in pleuropulmonary blastoma: a report from the International Pleuropulmonary Blastoma Registry. Pediatr Blood Cancer 56 (4): 604-9, 2011.
-
Ohta Y, Fujishima M, Hasegawa H, et al.: High therapeutic effectiveness of postoperative irinotecan chemotherapy in a typical case of radiographically and pathologically diagnosed pleuropulmonary blastoma. J Pediatr Hematol Oncol 31 (5): 355-8, 2009.
-
Venkatramani R, Malogolowkin MH, Wang L, et al.: Pleuropulmonary blastoma: a single-institution experience. J Pediatr Hematol Oncol 34 (5): e182-5, 2012.
-
de Castro CG Jr, de Almeida SG, Gregianin LJ, et al.: High-dose chemotherapy and autologous peripheral blood stem cell rescue in a patient with pleuropulmonary blastoma. J Pediatr Hematol Oncol 25 (1): 78-81, 2003.
-
MacSweeney F, Papagiannopoulos K, Goldstraw P, et al.: An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol 27 (8): 1139-46, 2003.
-
Hill DA, Dehner LP: A cautionary note about congenital cystic adenomatoid malformation (CCAM) type 4. Am J Surg Pathol 28 (4): 554-5; author reply 555, 2004.
-
Gangopadhyay AN, Mohanty PK, Gopal SC, et al.: Adenocarcinoma of the esophagus in an 8-year-old boy. J Pediatr Surg 32 (8): 1259-60, 1997.
-
Issaivanan M, Redner A, Weinstein T, et al.: Esophageal carcinoma in children and adolescents. J Pediatr Hematol Oncol 34 (1): 63-7, 2012.
-
Verley JM, Hollmann KH: Thymoma. A comparative study of clinical stages, histologic features, and survival in 200 cases. Cancer 55 (5): 1074-86, 1985.
-
Hsueh C, Kuo TT, Tsang NM, et al.: Thymic lymphoepitheliomalike carcinoma in children: clinicopathologic features and molecular analysis. J Pediatr Hematol Oncol 28 (12): 785-90, 2006.
-
Stachowicz-Stencel T, Bien E, Balcerska A, et al.: Thymic carcinoma in children: a report from the Polish Pediatric Rare Tumors Study. Pediatr Blood Cancer 54 (7): 916-20, 2010.
-
Carretto E, Inserra A, Ferrari A, et al.: Epithelial thymic tumours in paediatric age: a report from the TREP project. Orphanet J Rare Dis 6: 28, 2011.
-
Allan BJ, Thorson CM, Davis JS, et al.: An analysis of 73 cases of pediatric malignant tumors of the thymus. J Surg Res 184 (1): 397-403, 2013.
-
Fonseca AL, Ozgediz DE, Christison-Lagay ER, et al.: Pediatric thymomas: report of two cases and comprehensive review of the literature. Pediatr Surg Int 30 (3): 275-86, 2014.
-
Stachowicz-Stencel T, Orbach D, Brecht I, et al.: Thymoma and thymic carcinoma in children and adolescents: a report from the European Cooperative Study Group for Pediatric Rare Tumors (EXPeRT). Eur J Cancer 51 (16): 2444-52, 2015.
-
Souadjian JV, Enriquez P, Silverstein MN, et al.: The spectrum of diseases associated with thymoma. Coincidence or syndrome? Arch Intern Med 134 (2): 374-9, 1974.
-
Coulter D, Gold S: Thymoma in the offspring of a patient with Isaacs syndrome. J Pediatr Hematol Oncol 29 (11): 797-8, 2007.
-
Cowen D, Richaud P, Mornex F, et al.: Thymoma: results of a multicentric retrospective series of 149 non-metastatic irradiated patients and review of the literature. FNCLCC trialists. Fédération Nationale des Centres de Lutte Contre le Cancer. Radiother Oncol 34 (1): 9-16, 1995.
-
Tomaszek S, Wigle DA, Keshavjee S, et al.: Thymomas: review of current clinical practice. Ann Thorac Surg 87 (6): 1973-80, 2009.
-
Niehues T, Harms D, Jürgens H, et al.: Treatment of pediatric malignant thymoma: long-term remission in a 14-year-old boy with EBV-associated thymic carcinoma by aggressive, combined modality treatment. Med Pediatr Oncol 26 (6): 419-24, 1996.
-
Casey EM, Kiel PJ, Loehrer PJ Sr: Clinical management of thymoma patients. Hematol Oncol Clin North Am 22 (3): 457-73, 2008.
-
Giaccone G, Ardizzoni A, Kirkpatrick A, et al.: Cisplatin and etoposide combination chemotherapy for locally advanced or metastatic thymoma. A phase II study of the European Organization for Research and Treatment of Cancer Lung Cancer Cooperative Group. J Clin Oncol 14 (3): 814-20, 1996.
-
Loehrer PJ Sr, Jiroutek M, Aisner S, et al.: Combined etoposide, ifosfamide, and cisplatin in the treatment of patients with advanced thymoma and thymic carcinoma: an intergroup trial. Cancer 91 (11): 2010-5, 2001.
-
Carlson RW, Dorfman RF, Sikic BI: Successful treatment of metastatic thymic carcinoma with cisplatin, vinblastine, bleomycin, and etoposide chemotherapy. Cancer 66 (10): 2092-4, 1990.
-
Ströbel P, Bargou R, Wolff A, et al.: Sunitinib in metastatic thymic carcinomas: laboratory findings and initial clinical experience. Br J Cancer 103 (2): 196-200, 2010.
-
Butany J, Nair V, Naseemuddin A, et al.: Cardiac tumours: diagnosis and management. Lancet Oncol 6 (4): 219-28, 2005.
-
Bielefeld KJ, Moller JH: Cardiac tumors in infants and children: study of 120 operated patients. Pediatr Cardiol 34 (1): 125-8, 2013.
-
Burke A, Virmani R: Pediatric heart tumors. Cardiovasc Pathol 17 (4): 193-8, 2008 Jul-Aug.
-
Becker AE: Primary heart tumors in the pediatric age group: a review of salient pathologic features relevant for clinicians. Pediatr Cardiol 21 (4): 317-23, 2000 Jul-Aug.
-
Padalino MA, Vida VL, Boccuzzo G, et al.: Surgery for primary cardiac tumors in children: early and late results in a multicenter European Congenital Heart Surgeons Association study. Circulation 126 (1): 22-30, 2012.
-
Isaacs H Jr: Fetal and neonatal cardiac tumors. Pediatr Cardiol 25 (3): 252-73, 2004 May-Jun.
-
Elderkin RA, Radford DJ: Primary cardiac tumours in a paediatric population. J Paediatr Child Health 38 (2): 173-7, 2002.
-
Uzun O, Wilson DG, Vujanic GM, et al.: Cardiac tumours in children. Orphanet J Rare Dis 2: 11, 2007.
-
Bruce CJ: Cardiac tumours: diagnosis and management. Heart 97 (2): 151-60, 2011.
-
Boikos SA, Stratakis CA: Carney complex: the first 20 years. Curr Opin Oncol 19 (1): 24-9, 2007.
-
Carney JA, Young WF: Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist 2: 6-21, 1992.
-
Stratakis CA, Kirschner LS, Carney JA: Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86 (9): 4041-6, 2001.
-
Boikos SA, Stratakis CA: Carney complex: pathology and molecular genetics. Neuroendocrinology 83 (3-4): 189-99, 2006.
-
Kogon B, Shehata B, Katzenstein H, et al.: Primary congenital infantile fibrosarcoma of the heart: the first confirmed case. Ann Thorac Surg 91 (4): 1276-80, 2011.
-
Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013.
-
Kocabaş A, Ekici F, Cetin Iİ, et al.: Cardiac rhabdomyomas associated with tuberous sclerosis complex in 11 children: presentation to outcome. Pediatr Hematol Oncol 30 (2): 71-9, 2013.
-
Tworetzky W, McElhinney DB, Margossian R, et al.: Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol 92 (4): 487-9, 2003.
-
Bader RS, Chitayat D, Kelly E, et al.: Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr 143 (5): 620-4, 2003.
-
Neri M, Di Donato S, Maglietta R, et al.: Sudden death as presenting symptom caused by cardiac primary multicentric left ventricle rhabdomyoma, in an 11-month-old baby. An immunohistochemical study. Diagn Pathol 7: 169, 2012.
-
Beroukhim RS, Prakash A, Buechel ER, et al.: Characterization of cardiac tumors in children by cardiovascular magnetic resonance imaging: a multicenter experience. J Am Coll Cardiol 58 (10): 1044-54, 2011.
-
Michler RE, Goldstein DJ: Treatment of cardiac tumors by orthotopic cardiac transplantation. Semin Oncol 24 (5): 534-9, 1997.
-
Stiller B, Hetzer R, Meyer R, et al.: Primary cardiac tumours: when is surgery necessary? Eur J Cardiothorac Surg 20 (5): 1002-6, 2001.
-
Günther T, Schreiber C, Noebauer C, et al.: Treatment strategies for pediatric patients with primary cardiac and pericardial tumors: a 30-year review. Pediatr Cardiol 29 (6): 1071-6, 2008.
-
Wu KH, Mo XM, Liu YL: Clinical analysis and surgical results of cardiac myxoma in pediatric patients. J Surg Oncol 99 (1): 48-50, 2009.
-
Kutluk T, Demir HA, Büyükpamukçu M, et al.: Cardiac rhabdomyomas in childhood: six cases from a single institution. Turk J Pediatr 55 (1): 69-73, 2013 Jan-Feb.
-
Choudhry S, Nguyen HH, Anwar S: Rapid resolution of cardiac rhabdomyomas following everolimus therapy. BMJ Case Rep 2015: , 2015.
-
Nagata S, Nakanishi R: Malignant pleural mesothelioma with cavity formation in a 16-year-old boy. Chest 127 (2): 655-7, 2005.
-
Rosas-Salazar C, Gunawardena SW, Spahr JE: Malignant pleural mesothelioma in a child with ataxia-telangiectasia. Pediatr Pulmonol 48 (1): 94-7, 2013.
-
Hofmann J, Mintzer D, Warhol MJ: Malignant mesothelioma following radiation therapy. Am J Med 97 (4): 379-82, 1994.
-
Pappo AS, Santana VM, Furman WL, et al.: Post-irradiation malignant mesothelioma. Cancer 79 (1): 192-3, 1997.
-
Hyers TM, Ohar JM, Crim C: Clinical controversies in asbestos-induced lung diseases. Semin Diagn Pathol 9 (2): 97-101, 1992.
-
Kelsey A: Mesothelioma in childhood. Pediatr Hematol Oncol 11 (5): 461-2, 1994 Sep-Oct.
-
Moran CA, Albores-Saavedra J, Suster S: Primary peritoneal mesotheliomas in children: a clinicopathological and immunohistochemical study of eight cases. Histopathology 52 (7): 824-30, 2008.
-
Cioffredi LA, Jänne PA, Jackman DM: Treatment of peritoneal mesothelioma in pediatric patients. Pediatr Blood Cancer 52 (1): 127-9, 2009.
-
Maziak DE, Gagliardi A, Haynes AE, et al.: Surgical management of malignant pleural mesothelioma: a systematic review and evidence summary. Lung Cancer 48 (2): 157-69, 2005.
-
Krug LM, Pass HI, Rusch VW, et al.: Multicenter phase II trial of neoadjuvant pemetrexed plus cisplatin followed by extrapleural pneumonectomy and radiation for malignant pleural mesothelioma. J Clin Oncol 27 (18): 3007-13, 2009.
-
Treasure T: What is the best approach for surgery of malignant pleural mesothelioma? It is to put our efforts into obtaining trustworthy evidence for practice. J Thorac Cardiovasc Surg 151 (2): 307-9, 2016.
-
Milano E, Pourroy B, Rome A, et al.: Efficacy of a combination of pemetrexed and multiple redo-surgery in an 11-year-old girl with a recurrent multifocal abdominal mesothelioma. Anticancer Drugs 17 (10): 1231-4, 2006.
-
Sugalski A, Davis M, Prasannan L, et al.: Clinical, histologic, and genetic features of mesothelioma in a 7-year-old child. Pediatr Blood Cancer 60 (1): 146-8, 2013.
-
Kobayashi S, Waragai T, Sano H, et al.: Malignant peritoneal mesothelioma in a child: chemotherapy with gemcitabine and platinum was effective for the disease unresponsive to other treatments. Anticancer Drugs 25 (9): 1102-5, 2014.
-
Wall JE, Mandrell BN, Jenkins JJ 3rd, et al.: Effectiveness of paclitaxel in treating papillary serous carcinoma of the peritoneum in an adolescent. Am J Obstet Gynecol 172 (3): 1049-52, 1995.
Abdominal CancersAbdominal cancers include the following: - Adrenocortical carcinoma (carcinoma of the adrenal cortex).
- Gastric (stomach) cancer.
- Cancer of the pancreas.
- Colorectal carcinomas.
- Neuroendocrine tumors (carcinoid tumors).
- Gastrointestinal stromal tumors (GIST).
The prognosis, diagnosis, classification, and treatment of these abdominal cancers are discussed below. It must be emphasized that these cancers are seen very infrequently in patients younger than 15 years, and most of the evidence is derived from case series. (Refer to the PDQ summary on Wilms Tumor and Other Childhood Kidney Tumors for information about kidney tumors.) Adrenocortical carcinoma Adrenocortical tumors encompass a spectrum of diseases with often seamless transition from benign (adenoma) to malignant (carcinoma) behavior. Incidence The incidence of adrenocortical tumors in children is extremely low (only 0.2% of pediatric cancers).[1] Adrenocortical tumors appear to follow a bimodal distribution, with peaks during the first and fourth decades.[2,3] Childhood adrenocortical tumors typically present during the first 5 years of life (median age, 3-4 years), although there is a second, smaller peak during adolescence.[4,5,6,7,8,9] In children, 25 new cases are expected to occur annually in the United States, for an estimated annual incidence of 0.2 to 0.3 cases per 1 million individuals.[10] Internationally, however, the incidence of adrenocortical tumors appears to vary substantially. It is particularly high in southern Brazil, where it is approximately 10 to 15 times that observed in the United States.[11,12,13,14] Female gender is consistently predominant in most studies, with a female to male ratio of 1.6 to 1.0.[8,9,15] Risk Factors Germline TP53 mutations are almost always the predisposing factor. The likelihood of a TP53 germline mutation is highest in the first years of life and diminishes with age. Predisposing genetic factors have been implicated in more than 50% of the cases in North America and Europe and in 95% of the Brazilian cases. [16] - In the non-Brazilian cases, relatives of children with adrenocortical tumors often, although not invariably, have a high incidence of other non-adrenal cancers (Li-Fraumeni syndrome); germline mutations usually occur within the region coding for the TP53 DNA-binding domain (exons 5 to 8, primarily at highly conserved amino acid residues).[13,16]
- In the Brazilian cases, the patients' families do not exhibit a high incidence of cancer, and a single, unique mutation at codon 337 in exon 10 of the TP53 gene is consistently observed.[14,17] In a Brazilian study, neonatal screening for the TP53 R337H mutation, which is prevalent in the region, identified 461 (0.27%) carriers among 171,649 of the newborns who were screened.[18] Carriers and relatives younger than 15 years were offered clinical screening. Adrenocortical tumors identified in the screening participants were smaller and more curable than the tumors found in carriers who did not elect to participate in screening.
Patients with Beckwith-Wiedemann and hemihypertrophy syndromes have a predisposition to cancer, and as many as 16% of their neoplasms are adrenocortical tumors.[19] Hypomethylation of the KCNQ1OT1 gene has also been associated with the development of adrenocortical tumors in patients without the phenotypic features of Beckwith-Wiedemann syndrome.[20] However, less than 1% of children with adrenocortical tumors have these syndromes.[21] The distinctive genetic features of pediatric adrenocortical carcinoma have been reviewed.[22] Histology Unlike adult adrenocortical tumors, histologic differentiation of pediatric adenomas and carcinomas is difficult. However, approximately 10% to 20% of pediatric cases are adenomas.[2,5] The distinction between benign (adenomas) and malignant (carcinomas) tumors can be problematic. In fact, adenomas and carcinomas appear to share multiple genetic aberrations and may represent points on a continuum of cellular transformation.[23] Macroscopically, adenomas tend to be well defined and spherical, and they never invade surrounding structures. They are typically small (usually <200 cm3), and some studies have included size as a criterion for adenoma. By contrast, carcinomas have macroscopic features suggestive of malignancy; they are larger, and they show marked lobulation with extensive areas of hemorrhage and necrosis. Microscopically, carcinomas comprise larger cells with eosinophilic cytoplasm, arranged in alveolar clusters. Several authors have proposed histologic criteria that may help to distinguish the two types of neoplasm.[24,25] Morphologic criteria may not allow reliable distinction of benign and malignant adrenocortical tumors. Mitotic rate is consistently reported as the most important determinant of aggressive behavior.[26]IGF2 expression also appears to discriminate between carcinomas and adenomas in adults, but not in children.[27,28] Other histopathologic variables are also important, and risk groups may be identified on the basis of a score derived from tumor characteristics, such as tumor necrosis, mitotic rate, the presence of atypical mitoses, and venous, capsular, or adjacent organ invasion.[14,26] Biological Features A study performed on 71 pediatric adrenocortical tumors (37 in a discovery cohort and 34 in an independent cohort) provided a description of the genomic landscape of pediatric adrenocortical carcinoma.[29] - The most common genomic alteration, present in approximately 90% of cases, was copy number loss of heterozygosity for 11p15 with retention of the paternal allele resulting in IGF2 overexpression.
- TP53 mutations were commonly observed. Twelve of 71 cases had the Brazilian founder R337H TP53 germline mutation. Excluding the Brazilian founder mutation cases, TP53 germline mutations were observed in approximately one-third of cases, with somatic TP53 mutations observed in approximately 10% of the remaining cases, such that approximately 40% of non-Brazilian cases had TP53 mutations. Among cases with TP53 mutations, chromosome 17 loss of heterozygosity with selection against wild-type TP53 was present in virtually all cases.
- ATRX genomic alterations (primarily structural variants) were present in approximately 20% of cases. All ATRX alterations occurred in the presence of TP53 alterations. The co-occurrence of TP53 and ATRX mutations correlated with advanced stage, large tumor size, increased telomere length, and poor prognosis.
- Activating CTNNB1 mutations were found in approximately 20% of cases and were mutually exclusive with TP53 germline alterations.
Clinical Presentation Because pediatric adrenocortical tumors are almost universally functional, they cause endocrine disturbances, and a diagnosis is usually made 5 to 8 months after the first signs and symptoms emerge.[3,5] - Virilization (pubic hair, accelerated growth, enlarged penis, clitoromegaly, hirsutism, and acne) caused by an excess of androgen secretion is seen, alone or in combination with hypercortisolism, in more than 80% of patients.[14,30]
- Hyperestrogenism can also occur.[31]
- Isolated Cushing syndrome is very rare (5% of patients), and it appears to occur more frequently in older children.[3,5,8,14,32]
Because of the hormone hypersecretion, it is possible to establish an endocrine profile for each particular tumor, which may facilitate the evaluation of response to treatment and monitor for tumor recurrence.[14] Nonfunctional tumors are rare (<10%) and tend to occur in older children.[3] Prognostic Factors In patients with localized disease, younger age (<4 years), virilization alone, normal blood pressure, disease stage I, absence of spillage during surgery, and tumor weight no greater than 200 grams are associated with a greater probability of survival.[9,33] In a Cox regression model analysis, only stage I, virilization alone, and age 0 to 3 years were independently associated with a better outcome.[3] Available data suggest that tumor size is especially important in children; patients with small tumors have an excellent outcome with surgery alone, regardless of histologic features.[9,34] A European review of 82 patients (62 with localized disease, 20 with metastases) found that tumor volume of higher than 200 cm3, incomplete excision, age older than 5 years, and the presence of two or more risk factors were adverse prognostic factors. Overall survival (OS) at 3 years was 55% for the whole population and 73% for patients with localized disease; all patients with metastases died within 15 months of diagnosis. When the 62 patients with localized disease were analyzed separately, tumor volume of higher than 200 cm3 was a significant prognostic factor for progression-free survival (PFS) (hazard ratio [HR], 4.38) and OS (HR, 3.68).[35] A low expression of the HLA class II antigens HLA-DRA, HLA-DPA1, and HLA-DPB1 has been associated with older age, larger tumor size, presence of metastatic disease, and worse outcome.[36] The overall probability of 5-year survival for children with adrenocortical tumors is reported to be 54% to 74%.[3,5,6,8,32,33,34,35] Treatment At the time of diagnosis, two-thirds of pediatric patients have limited disease (tumors can be completely resected), and the remaining patients have either unresectable or metastatic disease.[3] Treatment of childhood adrenocortical tumors has evolved from the data derived from the adult studies, and the same guidelines are used. Surgery is the most important mode of therapy, and mitotane and cisplatin-based regimens, usually incorporating doxorubicin and etoposide, are recommended for patients with advanced disease.[13,14,37,38]; [8][Level of evidence: 3iiiA] Treatment options for childhood adrenocortical tumors include the following: - Surgery: An aggressive surgical approach toward the primary tumor and all metastatic sites is recommended when feasible.[39,40] Because of tumor friability, rupture of the capsule with resultant tumor spillage is frequent (approximately 20% of initial resections and 43% of resections after recurrence).[3,6] When the diagnosis of adrenocortical tumor is suspected, laparotomy and a curative procedure are recommended rather than fine-needle aspiration, to avoid the risk of tumor rupture.[40,41] Laparoscopic resection is associated with a high risk of rupture and peritoneal carcinomatosis; thus, open adrenalectomy remains the standard of care.[42]
- Mitotane and cisplatin-based regimens: Little information is available about the use of mitotane in children, although response rates appear to be similar to those seen in adults.[1,37] In adults, mitotane is commonly used as a single agent in the adjuvant setting after complete resection.[37]
- A retrospective analysis in Italy and Germany identified 177 adult patients with completely resected adrenocortical carcinoma. Recurrence-free survival was significantly prolonged by the use of adjuvant mitotane. Benefit was present with 1 g to 3 g per day of mitotane and was associated with fewer toxic side effects than doses of 3 g to 5 g per day.[43] (Refer to the PDQ summary on adult Adrenocortical Carcinoma Treatment for more information.)
- In a review of 11 children with advanced adrenocortical tumors treated with mitotane and a cisplatin-based chemotherapeutic regimen, measurable responses were seen in seven patients. The mitotane daily dose required for therapeutic levels was around 4 g/m2, and therapeutic levels were achieved after 4 to 6 months of therapy.[37]
- In the GPOH-MET 97 trial, mitotane levels greater than 14 mg/L correlated with better survival.[8,14]
The use of radiation therapy in pediatric patients with adrenocortical tumors has not been consistently investigated. Adrenocortical tumors are generally considered to be radioresistant. Furthermore, because many children with adrenocortical tumors carry germline TP53 mutations that predispose to cancer, radiation may increase the incidence of secondary tumors. One study reported that three of five long-term survivors of pediatric adrenocortical tumors died of secondary sarcoma that arose within the radiation field.[14,44] (Refer to the PDQ summary on adult Adrenocortical Carcinoma Treatment for more information.) Gastric (Stomach) Cancer Incidence Primary gastric tumors in children are rare, and carcinoma of the stomach is even more unusual.[45] In one series, gastric cancer in children younger than 18 years accounted for 0.11% of all gastric cancer cases seen over an 18-year period.[46] The frequency and death rate from stomach cancer has declined worldwide for the past 50 years with the introduction of food preservation practices such as refrigeration.[47] Clinical Presentation and Diagnostic Evaluation The tumor must be distinguished from other conditions such as non-Hodgkin lymphoma, malignant carcinoid, leiomyosarcoma, and various benign conditions or tumors of the stomach.[45] Symptoms of carcinoma of the stomach include the following: - Vague upper abdominal pain, which can be associated with poor appetite and weight loss.
- Nausea and vomiting.
- Change in bowel habits.
- Poor appetite.
- Weakness.
- Helicobacter pylori infection.[46,48]
- Anemia. Many individuals become anemic but otherwise show no symptoms before the development of metastatic spread.
Fiberoptic endoscopy can be used to visualize the tumor or to take a biopsy sample to confirm the diagnosis. Confirmation can also involve an x-ray examination of the upper gastrointestinal tract. Treatment and Outcome Treatment includes surgical excision with wide margins. For individuals who cannot have a complete surgical resection, radiation therapy may be used along with chemotherapeutic agents such as fluorouracil (5-FU) and irinotecan.[49] Other agents that may be of value are the nitrosoureas with or without cisplatin, etoposide, doxorubicin, or mitomycin C. Prognosis depends on the extent of the disease at the time of diagnosis and the success of treatment that is appropriate for the clinical situation.[46] Because of the rarity of stomach cancer in the pediatric age group, little information exists regarding the treatment outcomes of children. (Refer to the PDQ summary on adult Gastric Cancer Treatment for more information.) Cancer of the Pancreas Malignant pancreatic tumors are rare in children and adolescents, with an incidence of 0.46 cases per 1 million individuals younger than 30 years.[50,51,52,53] The primary pancreatic tumors of childhood can be classified into the following four categories: - Solid pseudopapillary tumor of the pancreas.
- Pancreatoblastoma.
- Islet cell tumors.
- Pancreatic carcinoma.
Solid Pseudopapillary Tumor of the Pancreas Solid pseudopapillary tumor of the pancreas, also known as Frantz tumor, is the most common pediatric pancreatic tumor, accounting for up to 70% of cases in most institutional series.[52,54] This tumor has low malignant potential and most commonly affects females of reproductive age (median age, 21 years), with a predilection for blacks and East Asians.[50,52,55] There is no known genetic or hormonal factor to explain the strong female predilection, although it has been noted that all tumors express progesterone receptors.[56] Solid pseudopapillary tumor of the pancreas is a very friable tumor, and tumor rupture and hemoperitoneum have been reported.[50,52,55] Tumors can occur throughout the pancreas and are often exophytic. On imaging, the mass shows typical cystic and solid components, with intratumoral hemorrhage and a fibrous capsule.[50] Histologically, the tumors are characterized by a combination of solid, pseudopapillary, and cystic changes. The fragility of the vascular supply leads to secondary degenerative changes and cystic areas of hemorrhage and necrosis. The cells surrounding the hyalinized fibrovascular stalks form the pseudopapillae.[50] A highly specific paranuclear dot-like immunoreactivity pattern for CD99 has been described.[57] The outcome of solid pseudopapillary tumors of the pancreas is excellent, with 10-year survival rates in excess of 95%.[56] Treatment of solid pseudopapillary tumor of the pancreas is surgical; however, preoperative and operative spillage is not unusual. Surgery is usually curative, although local recurrences occur in 5% to 15% of the cases.[55] Metastatic disease, usually in the liver, may occur in up to 15% of the cases.[50,52,55,56,57] Single-agent gemcitabine has been reported to be effective in cases of unresectable or metastatic disease.[58] Pancreatoblastoma Pancreatoblastoma accounts for 10% to 20% of all pancreatic tumors during childhood. It is the most common pancreatic tumor of young children and typically presents in the first decade of life, with a median age at diagnosis of 5 years.[50,59] Patients with Beckwith-Wiedemann syndrome have an increased risk of developing pancreatoblastoma; this syndrome is identified in up to 60% of cases of pancreatoblastoma developing during early infancy and in 5% of children developing pancreatoblastoma later in life.[60] Pancreatoblastoma has also been associated with familial adenomatous polyposis syndromes.[61] This tumor is thought to arise from the persistence of the fetal analog of pancreatic acinar cells. Pathology shows an epithelial neoplasm with an arrangement of acinar, trabecular, or solid formations separated by dense stromal bands.[50]CTNNB1 gene mutations have been described in some cases, suggesting that pancreatoblastoma might result from alterations in the normal pancreas differentiation.[62] Although approximately one-half of the cases originate in the head of the pancreas, jaundice is uncommon. Close to 80% of the tumors secrete alpha-fetoprotein, which can be used to measure response to therapy and monitor for recurrence.[59] In some cases, the tumor may secrete adrenocorticotropic hormone (ACTH) or antidiuretic hormone, and patients may present with Cushing syndrome and syndrome of inappropriate antidiuretic hormone secretion.[60] Metastases are present in 30% to 40% of the patients, usually involving liver, lungs, and lymph nodes.[59] Using a multimodality approach, close to 80% of patients can be cured.[59] Surgery is the mainstay in the treatment of pancreatoblastoma, and a complete surgical resection is required for cure. Because of the common origin in the head of the pancreas, a Whipple procedure is usually required. For large, unresectable, or metastatic tumors, preoperative chemotherapy is indicated; pancreatoblastoma commonly responds to chemotherapy, and a cisplatin-based regimen is usually recommended. The PLADO regimen, which includes cisplatin and doxorubicin, is the most commonly used regimen, and treatment is modeled after the management of hepatoblastoma, with two to three cycles of preoperative therapy, followed by resection and adjuvant chemotherapy.[52,59,61,63] Although radiation therapy has been used in unresectable or relapsed cases, its role in the treatment of microscopic disease after surgery has not been defined.[61] Response has been seen for patients with relapsed or persistent pancreatoblastoma treated with gemcitabine in one case [64] and vinorelbine and oral cyclophosphamide in two cases.[65] High-dose chemotherapy with autologous hematopoietic stem cell rescue has been reported to be effective in selected cases.[52,66] Islet Cell Tumors Islet cell tumors represent approximately 15% of pediatric pancreatic tumors in most series.[52,54,67] These tumors usually present in middle age and may be associated with multiple endocrine neoplasia type 1 (MEN1) syndrome; less than 5% of islet cell tumors occur in children.[50] The most common type of functioning islet cell tumor is insulinoma, followed by gastrinoma. Patients with insulinoma present with fasting hyperinsulinic hypoglycemia; in young children, presentation may include behavioral problems, seizures, or coma. Gastrinoma presents with Zollinger-Ellison syndrome, with recurrent peptic ulcers in uncommon locations, and diarrhea due to gastric hypersecretion. While most insulinomas are benign, a significant proportion of gastrinomas are malignant.[67] Other less common tumors seldom seen in children are the ACTHoma, which presents as Cushing syndrome, and the VIPoma, which presents as Verner-Morrison syndrome. Nonfunctioning tumors are extremely rare in pediatrics, except when associated with MEN1. Islet cell tumors are typically solitary; when multiple tumors are present, the diagnosis of MEN1 syndrome should be considered. On imaging, these tumors are usually small and well defined. Somatostatin receptor scintigraphy is useful for the location of islet cell tumors; however, only 60% to 70% express somatostatin receptor.[50] Treatment of islet cell tumors includes medical therapy for control of the syndrome and complete surgical resection. For patients with malignant tumors and unresectable or metastatic disease, chemotherapy and mammalian target of rapamycin (mTOR) inhibitors are recommended. The management of these tumors in children follows the consensus guidelines established for adult patients.[67,68] (Refer to the PDQ summary on adult Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment for more information.) Pancreatic Carcinoma Pancreatic carcinomas (acinar cell carcinoma and ductal adenocarcinoma) are extremely rare in children. These malignancies represent less than 5% of pediatric pancreatic tumors and include the following:[52,54] - Acinar cell carcinoma. Although rare in pediatrics, acinar cell carcinoma is more common than ductal cell adenocarcinoma, the most common pancreatic carcinoma in adults. Acinar cell carcinoma is considered to be the adult counterpart of pancreatoblastoma, and histological differentiation between both entities may be difficult.[50]
- Ductal adenocarcinoma. Ductal adenocarcinoma is extremely rare in the first four decades of life. However, ductal adenocarcinoma is associated with several cancer predisposition syndromes, such as hereditary pancreatitis (PRSS1 mutations), familial atypical mole and multiple melanoma (CDKN2 mutations), Peutz-Jeghers syndrome and other hereditary non-polyposis colon carcinomas (STK11 and germline mismatch repair genes), and syndromes associated with DNA repair gene mutations (such as BRCA2 and ATM).[69] Age at presentation may be younger in these patients, although occurrence during childhood and adolescence is extremely rare.[70]
Presenting symptoms are nonspecific and are related to local tumor growth. However, 4% to 15% of adult patients with acinar cell carcinoma may present with a lipase hypersecretion syndrome, manifesting as peripheral polyarthropathy and painful subcutaneous nodules. (Refer to the PDQ summary on adult Pancreatic Cancer Treatment for information about the treatment of pancreatic carcinoma.) Colorectal Carcinoma Incidence Carcinoma of the large bowel is rare in the pediatric age group.[71] It is seen in one case per 1 million persons younger than 20 years in the United States annually; fewer than 100 cases are diagnosed in children each year in the United States.[72] From 1973 to 2006, the Surveillance, Epidemiology, and End Results database recorded 174 cases of colorectal cancer in patients younger than 19 years.[73] Clinical Presentation Colorectal tumors can occur in any location in the large bowel. Larger series and reviews suggest that ascending and descending colon tumors are each seen in approximately 30% of cases, with rectal tumors occurring in approximately 25% of cases.[74,75,76] Signs and symptoms in children with descending colon tumors include the following: - Abdominal pain (most common).
- Rectal bleeding.
- Change in bowel habits.
- Weight loss.
- Nausea and vomiting.
The median duration of symptoms before diagnosis was about 3 months in one series.[72,77] Changes in bowel habits may be associated with tumors of the rectum or lower colon. Tumors of the right colon may cause more subtle symptoms but are often associated with the following: - Abdominal mass.
- Weight loss.
- Decreased appetite.
- Blood in the stool
- Iron-deficiency anemia.
Any tumor that causes complete obstruction of the large bowel can cause bowel perforation and spread of the tumor cells within the abdominal cavity. Diagnostic Evaluation Diagnostic studies include the following:[78,79] - Examination of the stool for blood.
- Studies of liver and kidney function.
- Measurement of carcinoembryonic antigen.
- Various medical imaging studies, including direct examination using colonoscopy to detect polyps in the large bowel. Other conventional radiographic studies include barium enema or video-capsule endoscopy followed by computed tomography of the chest and bone scans.[80]
Histology There is a higher incidence of mucinous adenocarcinoma in the pediatric and adolescent age group (40%-50%), with many lesions being the signet ring cell type,[71,72,77,81] whereas only about 15% of adult lesions are of this histology. The tumors of younger patients with this histologic variant may be less responsive to chemotherapy. In the adolescent and young adult population with the mucinous histology, there is a higher incidence of signet ring cells, microsatellite instability, and mutations in the mismatch repair genes.[82] Tumors with mucinous histology arise from the surface of the bowel, usually at the site of an adenomatous polyp. The tumor may extend into the muscle layer surrounding the bowel, or the tumor may perforate the bowel entirely and seed through the spaces around the bowel, including intra-abdominal fat, lymph nodes, liver, ovaries, and the surface of other loops of bowel. A high incidence of metastasis involving the pelvis, ovaries, or both may be present in girls.[79] Colorectal cancers in younger patients with noninherited sporadic tumors often lack KRAS mutations and other cytogenetic anomalies seen in older patients.[83] Staging Most reports also suggest that children present with more advanced disease than do adults, with 80% to 90% of patients presenting with Dukes stage C/D or TNM stage III/IV disease (refer to the Stage Information for Colon Cancer section of the PDQ summary on adult Colon Cancer Treatment for more information about staging).[72,74,75,76,77,78,81,84,85,86,87,88,89,90] Treatment and Outcome Most patients present with evidence of metastatic disease,[77] either as gross tumor or as microscopic deposits in lymph nodes, on the surface of the bowel, or on intra-abdominal organs.[81,84] Treatment options for childhood colorectal cancer include the following: - Surgery: Complete surgical excision is the most important prognostic factor and is the primary goal of surgery, but in most instances, this is impossible. Removal of large portions of tumor provides little benefit for those with extensive metastatic disease.[72] Most patients with microscopic metastatic disease generally develop gross metastatic disease, and few individuals with metastatic disease at diagnosis become long-term survivors.
- Radiation therapy and chemotherapy: Current therapy includes the use of radiation for rectal and lower colon tumors, in conjunction with chemotherapy using 5-FU with leucovorin.[91] Other agents, including irinotecan, may be of value.[77][Level of evidence: 3iiiA] No significant benefit has been determined for interferon-alpha given in conjunction with 5-FU/leucovorin.[92]
A recent review of nine clinical trials comprising 138 patients younger than 40 years demonstrated that the use of combination chemotherapy improved PFS and OS in these patients. Furthermore, OS and response rates to chemotherapy were similar to those observed in older patients.[93][Level of evidence: 2A] Other active agents used in adults include oxaliplatin, bevacizumab, panitumumab, cetuximab, aflibercept, and regorafenib.[94,95,96,97]
Survival is consistent with the advanced stage of disease observed in most children with colorectal cancer, with an overall mortality rate of approximately 70%. For patients with a complete surgical resection or for those with low-stage/localized disease, survival is significantly prolonged, with the potential for cure.[74] Genetic Syndromes Associated With Colorectal Cancer About 20% to 30% of adult patients with colorectal cancer have a significant history of familial cancer; of these, about 5% have a well-defined genetic syndrome.[98] The incidence of these genetic syndromes in children has not been well defined, as follows: - In one review, 16% of patients younger than 40 years had a predisposing factor for the development of colorectal cancer.[99]
- A later study documented immunohistochemical evidence of mismatch repair deficiency in 31% of colorectal carcinoma samples in patients aged 30 years or younger.[100]
- A retrospective review of patients younger than 18 years in Germany identified 31 patients with colorectal carcinoma.[101] Eleven of the 26 patients who were tested for a genetic predisposition syndrome tested positive (eight cases of Lynch syndrome, one patient with familial adenomatous polyposis, and two patients with constitutional mismatch repair deficiency). When compared with the patients without a genetic predisposition syndrome, the 11 patients with a genetic predisposition syndrome presented with more localized disease, allowing complete surgical resection and improved outcome (100% survival).
The most common genetic syndromes associated with the development of colorectal cancer are shown in Tables 5 and 6. Table 5. Common Genetic Syndromes Associated With Adenomatous Polyposis| Syndrome | Gene | Gene Function | Hereditary Pattern |
|---|
| Attenuated familial adenomatous polyposis | APC(5' mutations),AXIN2 | Tumor suppressor | Dominant | | Familial adenomatous polyposis (Gardner syndrome) | APC | Tumor suppressor | Dominant | | Lynch syndrome (hereditary nonpolyposis colorectal cancer) | MSH2, MLH1, MSH6, PMS2, EPCAM | Repair/stability | Dominant | | Li-Fraumeni syndrome | TP53(p53) | Tumor suppressor | Dominant | | MYH-associated polyposis | MYH(MUTYH) | Repair/stability | Recessive | | Turcot syndrome | APC | Tumor suppressor | Dominant | | MLH1 | Repair/stability | Dominant | Table 6. Common Genetic Syndromes Associated With Hamartomatous Polyps| Syndrome | Gene | Gene Function | Hereditary Pattern |
|---|
| Cowden syndrome | PTEN | Tumor suppressor | Dominant | | Juvenile polyposis syndrome | BMPR1A, SMAD4, ENG | Tumor suppressor | Dominant | | Peutz-Jeghers syndrome | STK11 | Tumor suppressor | Dominant | Familial polyposis is inherited as a dominant trait, which confers a high degree of risk. Early diagnosis and surgical removal of the colon eliminates the risk of developing carcinomas of the large bowel.[102] Some colorectal carcinomas in young people, however, may be associated with a mutation of the adenomatous polyposis coli (APC) gene, which also is associated with an increased risk of brain tumors and hepatoblastoma.[103] Familial adenomatous polyposis (FAP) syndrome is caused by mutation of a gene on chromosome 5q, which normally suppresses proliferation of cells lining the intestine and later development of polyps.[104] A double-blind, placebo-controlled, randomized phase I trial in children aged 10 to 14 years with FAP reported that celecoxib at a dose of 16 mg/kg per day is safe for administration for up to 3 months. At this dose, there was a significant decrease in the number of polyps detected on colonoscopy.[105][Level of evidence: 1iiDiv] The role of celecoxib in the management of FAP in children is not clear. Another tumor suppressor gene on chromosome 18 is associated with progression of polyps to malignant form. Multiple colon carcinomas have been associated with neurofibromatosis type I and several other rare syndromes.[106] Despite the increased risk of multiple malignancies in families with Lynch syndrome, the risk of malignant neoplasms during childhood in those families does not seem to be increased when compared with the risk in children from non-Lynch syndrome colorectal carcinoma families.[107] (Refer to the PDQ summary on Genetics of Colorectal Cancer for more information about the genetic syndromes associated with childhood colorectal cancer.) Neuroendocrine Tumors (Carcinoid Tumors) These tumors, like bronchial adenomas, may be benign or malignant and can involve the lining of the lung, large or small bowel, or liver.[108,109,110,111,112,113] Most lung lesions are benign; however, some metastasize.[114] A single-institution retrospective review identified 45 cases of carcinoid tumors in children and adolescents between 2003 and 2016.[115][Level of evidence: 3iiDii] The most common primary site was the appendix (36 of 45 cases). No recurrences were observed among the patients with appendiceal primary tumors treated with appendectomy alone, which supports resection of the appendix without hemicolectomy as the procedure of choice. Extra-appendiceal primary tumors were associated with a higher risk of metastasis and recurrence. The carcinoid syndrome of excessive excretion of somatostatin is characterized by flushing, labile blood pressure, and metastatic spread of the tumor to the liver.[114] Symptoms may be lessened by giving somatostatin analogs, which are available in short-acting and long-acting forms.[116] Occasionally, carcinoids may produce ectopic ACTH and cause Cushing disease.[117] Neuroendocrine Tumors of the Appendix Most carcinoid tumors of the appendix are discovered incidentally at the time of appendectomy, and are small, low-grade, localized tumors; simple appendectomy is the therapy of choice.[118,119,120] For larger (>2 cm) tumors or tumors that have spread to local nodes, cecectomy or rarely, right hemicolectomy, is the usual treatment. It has become accepted practice to remove the entire right colon in patients with large carcinoid tumors of the appendix (>2 cm in diameter) or with tumors that have spread to the nodes; however, this practice remains controversial.[121] - The German Society of Pediatric Oncology and Hematology has maintained a registry of appendiceal neuroendocrine tumors since 1997. They reported on 237 children and adolescents.[122][Level of evidence: 3iiDii] A second surgery or lymph node sampling was performed in 60 patients; infiltration of lymph nodes was found in 9 of these 60 patients. The group recommended secondary right hemicolectomy in completely resected appendiceal neuroendocrine tumors only for tumors larger than 15 mm and local follow-up resection with lymph node sampling for incompletely removed tumors smaller than 15 mm. These recommendations are controversial.
- The Italian Rare Tumors in Pediatric Age (TREP) project performed a prospective registry study that evaluated 113 patients with appendiceal neuroendocrine tumors.[123][Level of evidence: 3iiiA] Primary re-excision was not recommended for completely excised tumors smaller than 2 cm except for microscopic/macroscopic residual tumor on the margins of the appendix, in which case cecum resection and pericecal node biopsy was recommended. Decisions about tumors larger than 2 cm were made at the discretion of the primary physicians. However, physicians were discouraged from performing right hemicolectomy unless margins were positive. At 41 months of follow-up, 113 of 113 patients were alive. Of the 113 patients, 108 had tumors smaller than 2 cm. Thirty-five patients had extension of tumor beyond the appendiceal well. Five tumors invaded the serosa, and 28 tumors invaded the periappendiceal fat. Margins were clear in 111 of 113 patients. The five patients with tumors larger than 2 cm did well. One patient had resection of the cecum without residual tumor found. Only one patient had a right hemicolectomy (tumor was <2 cm with clear margins, but an octreotide scan was possibly positive; no tumor was found). The study concluded that appendectomy alone should be considered curative for most cases of appendiceal neuroendocrine tumors. The procedure of choice is a resection of the appendix without the need for hemicolectomy.
Nonappendiceal Neuroendocrine Tumors Nonappendiceal neuroendocrine tumors in the abdomen can occur in the pancreas, stomach, and liver. The most common clinical presentation is an unknown primary site. Nonappendiceal neuroendocrine tumors are more likely to be larger, higher grade, or present with metastases.[124] Larger tumor size has been associated with a higher risk of recurrence.[115] In one retrospective, single-institution study, the 5-year relapse-free survival rate of nonappendiceal neuroendocrine tumors was 41% and the overall survival rate was 66%. Chemotherapy for these tumors was largely ineffective.[124] (Refer to the Bronchial tumors section of this summary for information about bronchial carcinoid tumors.) Metastatic Neuroendocrine Tumors Treatment of metastatic carcinoid tumors of the large bowel, pancreas, or stomach becomes more complicated and requires treatment similar to that given for adult high-grade neuroendocrine tumors. (Refer to the PDQ summary on adult Gastrointestinal Carcinoid Tumors for treatment options in patients with malignant carcinoid tumors.) Gastrointestinal Stromal Tumors (GIST) Incidence Gastrointestinal stromal tumors (GIST) are the most common mesenchymal neoplasms of the gastrointestinal tract in adults.[125] These tumors are rare in children.[126] Approximately 2% of all GIST occur in children and young adults.[127,128,129] In one series, pediatric GIST accounted for 2.5% of all pediatric nonrhabdomyosarcomatous soft tissue sarcomas.[130] Previously, these tumors were diagnosed as leiomyomas, leiomyosarcomas, and leiomyoblastomas. In pediatric patients, GIST are most commonly located in the stomach and almost exclusively affect adolescent females.[129,131,132] Histology and Molecular Features Histologically, pediatric GIST have a predominance of epithelioid or epithelioid/spindle cell morphology and, unlike adult GIST, their mitotic rate does not appear to accurately predict clinical behavior.[131,133] The majority of GIST in the pediatric age range have loss of the succinate dehydrogenase (SDH) complex and consequently, lack SDHB expression by immunohistochemistry.[134,135] In addition, these tumors have minimal large-scale chromosomal changes and overexpress the insulin-like growth factor 1 receptor.[136,137] Activating mutations of KIT and PDGFA, which are seen in 90% of adult GIST, are present in only a small fraction of pediatric GIST.[131,136,138] The lack of SDHB expression in most pediatric GIST implicates cellular respiration defects in the pathogenesis of this disease and supports the notion that this disease is better classified as SDH-deficient GIST. Furthermore, about 50% of patients with SDH-deficient GIST have germline mutations of the SDH complex, most commonly involving SDHA,[134] supporting the notion that SDH-deficient GIST is a cancer predisposition syndrome and testing of affected patients for constitutional mutations for the SDH complex should be considered.[139] A small percentage of SDH-deficient GIST lack somatic or germline mutations of the SDH complex and are characterized by SDHC promoter hypermethylation and gene silencing and are categorized as SDH epimutant tumors.[140] Clinical Features Most pediatric patients with GIST are diagnosed during the second decade of life with anemia-related gastrointestinal bleeding. In addition, pediatric GIST have a high propensity for multifocality (23%) and nodal metastases.[129,131,138] These features may account for the high incidence of local recurrence seen in this patient population. Despite these features, patients have an indolent course characterized by multiple recurrences and long survival.[138] SDH-deficient GIST can arise within the context of the following two syndromes:[131,141] - Carney triad. Carney triad is a syndrome characterized by the occurrence of GIST, lung chondromas, and paragangliomas. In addition, about 20% of patients have adrenal adenomas and 10% have esophageal leiomyomas. GIST are the most common (75%) presenting lesions in these patients. To date, no coding sequence mutations of KIT, PDGFR, or the succinate dehydrogenase (SDH) genes have been found in these patients.[129,141,142]
- Carney-Stratakis syndrome. Carney-Stratakis syndrome is characterized by paraganglioma and GIST caused by germline mutations of the SDH genes B, C, and D.[135,143]
Treatment Once the diagnosis of pediatric GIST is established, referral to medical centers with expertise in the treatment of GIST should be considered, and that all samples be evaluated for mutations of KIT (exons 9, 11, 13, 17), PDGFR (exons 12, 14, 18), and BRAF (V600E).[144,145] Treatment of GIST depends on whether a mutation is detected, as follows: - GIST with a KIT or PDGFR mutation: Pediatric patients who harbor KIT or PDGFR mutations are managed according to adult guidelines.
- SDH-deficient GIST: For most pediatric patients with SDH-deficient GIST, complete surgical resection of localized disease is recommended as long as it can be accomplished without significant morbidity (i.e., gastrectomy). When feasible, wedge resections are an acceptable surgical option. Because lymph node involvement is relatively common in younger patients, searching for overt or occult nodal involvement is encouraged. Given the indolent course of the disease in pediatric patients, it is reasonable to withhold extensive and mutilative surgeries and to carefully observe children with locally recurrent or unresectable asymptomatic disease.[126,131]
Responses to imatinib and sunitinib in pediatric patients with SDH-deficient GIST are uncommon and consist mainly of disease stabilization.[131,146,147] In a review of ten patients who were treated with imatinib mesylate, one patient experienced a partial response and three patients had stable disease.[131] In another study, sunitinib appeared to show more activity, with one partial response and five cases of stable disease in six children with imatinib-resistant GIST.[148] Unlike the adult recommendations, the use of adjuvant imatinib cannot be recommended in children with SDH-deficient GIST.[149]
References:
-
Ribeiro RC, Figueiredo B: Childhood adrenocortical tumours. Eur J Cancer 40 (8): 1117-26, 2004.
-
Wooten MD, King DK: Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer 72 (11): 3145-55, 1993.
-
Michalkiewicz E, Sandrini R, Figueiredo B, et al.: Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol 22 (5): 838-45, 2004.
-
Ribeiro RC, Sandrini Neto RS, Schell MJ, et al.: Adrenocortical carcinoma in children: a study of 40 cases. J Clin Oncol 8 (1): 67-74, 1990.
-
Wieneke JA, Thompson LD, Heffess CS: Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol 27 (7): 867-81, 2003.
-
Sandrini R, Ribeiro RC, DeLacerda L: Childhood adrenocortical tumors. J Clin Endocrinol Metab 82 (7): 2027-31, 1997.
-
Bugg MF, Ribeiro RC, Roberson PK, et al.: Correlation of pathologic features with clinical outcome in pediatric adrenocortical neoplasia. A study of a Brazilian population. Brazilian Group for Treatment of Childhood Adrenocortical Tumors. Am J Clin Pathol 101 (5): 625-9, 1994.
-
Redlich A, Boxberger N, Strugala D, et al.: Systemic treatment of adrenocortical carcinoma in children: data from the German GPOH-MET 97 trial. Klin Padiatr 224 (6): 366-71, 2012.
-
Gulack BC, Rialon KL, Englum BR, et al.: Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB). J Pediatr Surg 51 (1): 172-7, 2016.
-
Berstein L, Gurney JG: Carcinomas and other malignant epithelial neoplasms. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, Chapter 11, pp 139-148. Also available online. Last accessed April 04, 2017.
-
Figueiredo BC, Sandrini R, Zambetti GP, et al.: Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet 43 (1): 91-6, 2006.
-
Pianovski MA, Maluf EM, de Carvalho DS, et al.: Mortality rate of adrenocortical tumors in children under 15 years of age in Curitiba, Brazil. Pediatr Blood Cancer 47 (1): 56-60, 2006.
-
Rodriguez-Galindo C, Figueiredo BC, Zambetti GP, et al.: Biology, clinical characteristics, and management of adrenocortical tumors in children. Pediatr Blood Cancer 45 (3): 265-73, 2005.
-
Rodriguez-Galindo C: Adrenocortical tumors in children. In: Schneider DT, Brecht IB, Olson TA: Rare Tumors in Children and Adolescents. Berlin, Germany: Springer-Verlag, 2012, pp 436-44.
-
Michalkiewicz EL, Sandrini R, Bugg MF, et al.: Clinical characteristics of small functioning adrenocortical tumors in children. Med Pediatr Oncol 28 (3): 175-8, 1997.
-
Wasserman JD, Novokmet A, Eichler-Jonsson C, et al.: Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children's oncology group study. J Clin Oncol 33 (6): 602-9, 2015.
-
Ribeiro RC, Sandrini F, Figueiredo B, et al.: An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 98 (16): 9330-5, 2001.
-
Custódio G, Parise GA, Kiesel Filho N, et al.: Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol 31 (20): 2619-26, 2013.
-
Hoyme HE, Seaver LH, Jones KL, et al.: Isolated hemihyperplasia (hemihypertrophy): report of a prospective multicenter study of the incidence of neoplasia and review. Am J Med Genet 79 (4): 274-8, 1998.
-
Wijnen M, Alders M, Zwaan CM, et al.: KCNQ1OT1 hypomethylation: a novel disguised genetic predisposition in sporadic pediatric adrenocortical tumors? Pediatr Blood Cancer 59 (3): 565-6, 2012.
-
Steenman M, Westerveld A, Mannens M: Genetics of Beckwith-Wiedemann syndrome-associated tumors: common genetic pathways. Genes Chromosomes Cancer 28 (1): 1-13, 2000.
-
El Wakil A, Doghman M, Latre De Late P, et al.: Genetics and genomics of childhood adrenocortical tumors. Mol Cell Endocrinol 336 (1-2): 169-73, 2011.
-
Figueiredo BC, Stratakis CA, Sandrini R, et al.: Comparative genomic hybridization analysis of adrenocortical tumors of childhood. J Clin Endocrinol Metab 84 (3): 1116-21, 1999.
-
Weiss LM: Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol 8 (3): 163-9, 1984.
-
van Slooten H, Schaberg A, Smeenk D, et al.: Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer 55 (4): 766-73, 1985.
-
Stojadinovic A, Ghossein RA, Hoos A, et al.: Adrenocortical carcinoma: clinical, morphologic, and molecular characterization. J Clin Oncol 20 (4): 941-50, 2002.
-
Almeida MQ, Fragoso MC, Lotfi CF, et al.: Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab 93 (9): 3524-31, 2008.
-
West AN, Neale GA, Pounds S, et al.: Gene expression profiling of childhood adrenocortical tumors. Cancer Res 67 (2): 600-8, 2007.
-
Pinto EM, Chen X, Easton J, et al.: Genomic landscape of paediatric adrenocortical tumours. Nat Commun 6: 6302, 2015.
-
Gönç EN, Özön ZA, Cakır MD, et al.: Need for comprehensive hormonal workup in the management of adrenocortical tumors in children. J Clin Res Pediatr Endocrinol 6 (2): 68-73, 2014.
-
Ghazi AA, Mofid D, Salehian MT, et al.: Functioning adrenocortical tumors in children-secretory behavior. J Clin Res Pediatr Endocrinol 5 (1): 27-32, 2013.
-
Hanna AM, Pham TH, Askegard-Giesmann JR, et al.: Outcome of adrenocortical tumors in children. J Pediatr Surg 43 (5): 843-9, 2008.
-
McAteer JP, Huaco JA, Gow KW: Predictors of survival in pediatric adrenocortical carcinoma: a Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg 48 (5): 1025-31, 2013.
-
Klein JD, Turner CG, Gray FL, et al.: Adrenal cortical tumors in children: factors associated with poor outcome. J Pediatr Surg 46 (6): 1201-7, 2011.
-
Cecchetto G, Ganarin A, Bien E, et al.: Outcome and prognostic factors in high-risk childhood adrenocortical carcinomas: A report from the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Pediatr Blood Cancer 64 (6): , 2017.
-
Leite FA, Lira RC, Fedatto PF, et al.: Low expression of HLA-DRA, HLA-DPA1, and HLA-DPB1 is associated with poor prognosis in pediatric adrenocortical tumors (ACT). Pediatr Blood Cancer 61 (11): 1940-8, 2014.
-
Zancanella P, Pianovski MA, Oliveira BH, et al.: Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma: mitotane monitoring and tumor regression. J Pediatr Hematol Oncol 28 (8): 513-24, 2006.
-
Hovi L, Wikström S, Vettenranta K, et al.: Adrenocortical carcinoma in children: a role for etoposide and cisplatin adjuvant therapy? Preliminary report. Med Pediatr Oncol 40 (5): 324-6, 2003.
-
Stewart JN, Flageole H, Kavan P: A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg 39 (5): 759-63, 2004.
-
Hubertus J, Boxberger N, Redlich A, et al.: Surgical aspects in the treatment of adrenocortical carcinomas in children: data of the GPOH-MET 97 trial. Klin Padiatr 224 (3): 143-7, 2012.
-
Kardar AH: Rupture of adrenal carcinoma after biopsy. J Urol 166 (3): 984, 2001.
-
Gonzalez RJ, Shapiro S, Sarlis N, et al.: Laparoscopic resection of adrenal cortical carcinoma: a cautionary note. Surgery 138 (6): 1078-85; discussion 1085-6, 2005.
-
Terzolo M, Angeli A, Fassnacht M, et al.: Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 356 (23): 2372-80, 2007.
-
Driver CP, Birch J, Gough DC, et al.: Adrenal cortical tumors in childhood. Pediatr Hematol Oncol 15 (6): 527-32, 1998 Nov-Dec.
-
Curtis JL, Burns RC, Wang L, et al.: Primary gastric tumors of infancy and childhood: 54-year experience at a single institution. J Pediatr Surg 43 (8): 1487-93, 2008.
-
Subbiah V, Varadhachary G, Herzog CE, et al.: Gastric adenocarcinoma in children and adolescents. Pediatr Blood Cancer 57 (3): 524-7, 2011.
-
American Cancer Society: Cancer Facts and Figures-2000. Atlanta, Ga: American Cancer Society, 2000.
-
Rowland M, Drumm B: Helicobacter pylori infection and peptic ulcer disease in children. Curr Opin Pediatr 7 (5): 553-9, 1995.
-
Ajani JA: Current status of therapy for advanced gastric carcinoma. Oncology (Huntingt) 12 (8 Suppl 6): 99-102, 1998.
-
Chung EM, Travis MD, Conran RM: Pancreatic tumors in children: radiologic-pathologic correlation. Radiographics 26 (4): 1211-38, 2006 Jul-Aug.
-
Perez EA, Gutierrez JC, Koniaris LG, et al.: Malignant pancreatic tumors: incidence and outcome in 58 pediatric patients. J Pediatr Surg 44 (1): 197-203, 2009.
-
Dall'igna P, Cecchetto G, Bisogno G, et al.: Pancreatic tumors in children and adolescents: the Italian TREP project experience. Pediatr Blood Cancer 54 (5): 675-80, 2010.
-
Brecht IB, Schneider DT, Klöppel G, et al.: Malignant pancreatic tumors in children and young adults: evaluation of 228 patients identified through the Surveillance, Epidemiology, and End Result (SEER) database. Klin Padiatr 223 (6): 341-5, 2011.
-
Rojas Y, Warneke CL, Dhamne CA, et al.: Primary malignant pancreatic neoplasms in children and adolescents: a 20 year experience. J Pediatr Surg 47 (12): 2199-204, 2012.
-
Papavramidis T, Papavramidis S: Solid pseudopapillary tumors of the pancreas: review of 718 patients reported in English literature. J Am Coll Surg 200 (6): 965-72, 2005.
-
Estrella JS, Li L, Rashid A, et al.: Solid pseudopapillary neoplasm of the pancreas: clinicopathologic and survival analyses of 64 cases from a single institution. Am J Surg Pathol 38 (2): 147-57, 2014.
-
Laje P, Bhatti TR, Adzick NS: Solid pseudopapillary neoplasm of the pancreas in children: a 15-year experience and the identification of a unique immunohistochemical marker. J Pediatr Surg 48 (10): 2054-60, 2013.
-
Maffuz A, Bustamante Fde T, Silva JA, et al.: Preoperative gemcitabine for unresectable, solid pseudopapillary tumour of the pancreas. Lancet Oncol 6 (3): 185-6, 2005.
-
Bien E, Godzinski J, Dall'igna P, et al.: Pancreatoblastoma: a report from the European cooperative study group for paediatric rare tumours (EXPeRT). Eur J Cancer 47 (15): 2347-52, 2011.
-
Chisholm KM, Hsu CH, Kim MJ, et al.: Congenital pancreatoblastoma: report of an atypical case and review of the literature. J Pediatr Hematol Oncol 34 (4): 310-5, 2012.
-
Glick RD, Pashankar FD, Pappo A, et al.: Management of pancreatoblastoma in children and young adults. J Pediatr Hematol Oncol 34 (Suppl 2): S47-50, 2012.
-
Honda S, Okada T, Miyagi H, et al.: Spontaneous rupture of an advanced pancreatoblastoma: aberrant RASSF1A methylation and CTNNB1 mutation as molecular genetic markers. J Pediatr Surg 48 (4): e29-32, 2013.
-
Défachelles AS, Martin De Lassalle E, Boutard P, et al.: Pancreatoblastoma in childhood: clinical course and therapeutic management of seven patients. Med Pediatr Oncol 37 (1): 47-52, 2001.
-
Belletrutti MJ, Bigam D, Bhargava R, et al.: Use of gemcitabine with multi-stage surgical resection as successful second-line treatment of metastatic pancreatoblastoma. J Pediatr Hematol Oncol 35 (1): e7-10, 2013.
-
Dhamne C, Herzog CE: Response of Relapsed Pancreatoblastoma to a Combination of Vinorelbine and Oral Cyclophosphamide. J Pediatr Hematol Oncol 37 (6): e378-80, 2015.
-
Hamidieh AA, Jalili M, Khojasteh O, et al.: Autologous stem cell transplantation as treatment modality in a patient with relapsed pancreatoblastoma. Pediatr Blood Cancer 55 (3): 573-6, 2010.
-
Jensen RT, Cadiot G, Brandi ML, et al.: ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 95 (2): 98-119, 2012.
-
Kulke MH, Benson AB 3rd, Bergsland E, et al.: Neuroendocrine tumors. J Natl Compr Canc Netw 10 (6): 724-64, 2012.
-
Rustgi AK: Familial pancreatic cancer: genetic advances. Genes Dev 28 (1): 1-7, 2014.
-
Lüttges J, Stigge C, Pacena M, et al.: Rare ductal adenocarcinoma of the pancreas in patients younger than age 40 years. Cancer 100 (1): 173-82, 2004.
-
da Costa Vieira RA, Tramonte MS, Lopes LF: Colorectal carcinoma in the first decade of life: a systematic review. Int J Colorectal Dis 30 (8): 1001-6, 2015.
-
Saab R, Furman WL: Epidemiology and management options for colorectal cancer in children. Paediatr Drugs 10 (3): 177-92, 2008.
-
Ferrari A, Casanova M, Massimino M, et al.: Peculiar features and tailored management of adult cancers occurring in pediatric age. Expert Rev Anticancer Ther 10 (11): 1837-51, 2010.
-
Kaplan MA, Isikdogan A, Gumus M, et al.: Childhood, adolescents, and young adults (≤25 y) colorectal cancer: study of Anatolian Society of Medical Oncology. J Pediatr Hematol Oncol 35 (2): 83-9, 2013.
-
Kim G, Baik SH, Lee KY, et al.: Colon carcinoma in childhood: review of the literature with four case reports. Int J Colorectal Dis 28 (2): 157-64, 2013.
-
Sultan I, Rodriguez-Galindo C, El-Taani H, et al.: Distinct features of colorectal cancer in children and adolescents: a population-based study of 159 cases. Cancer 116 (3): 758-65, 2010.
-
Hill DA, Furman WL, Billups CA, et al.: Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J Clin Oncol 25 (36): 5808-14, 2007.
-
Pratt CB, Rao BN, Merchant TE, et al.: Treatment of colorectal carcinoma in adolescents and young adults with surgery, 5-fluorouracil/leucovorin/interferon-alpha 2a and radiation therapy. Med Pediatr Oncol 32 (6): 459-60, 1999.
-
Kauffman WM, Jenkins JJ 3rd, Helton K, et al.: Imaging features of ovarian metastases from colonic adenocarcinoma in adolescents. Pediatr Radiol 25 (4): 286-8, 1995.
-
Postgate A, Hyer W, Phillips R, et al.: Feasibility of video capsule endoscopy in the management of children with Peutz-Jeghers syndrome: a blinded comparison with barium enterography for the detection of small bowel polyps. J Pediatr Gastroenterol Nutr 49 (4): 417-23, 2009.
-
Ferrari A, Rognone A, Casanova M, et al.: Colorectal carcinoma in children and adolescents: the experience of the Istituto Nazionale Tumori of Milan, Italy. Pediatr Blood Cancer 50 (3): 588-93, 2008.
-
Tricoli JV, Seibel NL, Blair DG, et al.: Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst 103 (8): 628-35, 2011.
-
Bleyer A, Barr R, Hayes-Lattin B, et al.: The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer 8 (4): 288-98, 2008.
-
Chantada GL, Perelli VB, Lombardi MG, et al.: Colorectal carcinoma in children, adolescents, and young adults. J Pediatr Hematol Oncol 27 (1): 39-41, 2005.
-
Durno C, Aronson M, Bapat B, et al.: Family history and molecular features of children, adolescents, and young adults with colorectal carcinoma. Gut 54 (8): 1146-50, 2005.
-
Karnak I, Ciftci AO, Senocak ME, et al.: Colorectal carcinoma in children. J Pediatr Surg 34 (10): 1499-504, 1999.
-
LaQuaglia MP, Heller G, Filippa DA, et al.: Prognostic factors and outcome in patients 21 years and under with colorectal carcinoma. J Pediatr Surg 27 (8): 1085-9; discussion 1089-90, 1992.
-
Radhakrishnan CN, Bruce J: Colorectal cancers in children without any predisposing factors. A report of eight cases and review of the literature. Eur J Pediatr Surg 13 (1): 66-8, 2003.
-
Sharma AK, Gupta CR: Colorectal cancer in children: case report and review of literature. Trop Gastroenterol 22 (1): 36-9, 2001 Jan-Mar.
-
Taguchi T, Suita S, Hirata Y, et al.: Carcinoma of the colon in children: a case report and review of 41 Japanese cases. J Pediatr Gastroenterol Nutr 12 (3): 394-9, 1991.
-
Madajewicz S, Petrelli N, Rustum YM, et al.: Phase I-II trial of high-dose calcium leucovorin and 5-fluorouracil in advanced colorectal cancer. Cancer Res 44 (10): 4667-9, 1984.
-
Wolmark N, Bryant J, Smith R, et al.: Adjuvant 5-fluorouracil and leucovorin with or without interferon alfa-2a in colon carcinoma: National Surgical Adjuvant Breast and Bowel Project protocol C-05. J Natl Cancer Inst 90 (23): 1810-6, 1998.
-
Blanke CD, Bot BM, Thomas DM, et al.: Impact of young age on treatment efficacy and safety in advanced colorectal cancer: a pooled analysis of patients from nine first-line phase III chemotherapy trials. J Clin Oncol 29 (20): 2781-6, 2011.
-
Saltz LB, Clarke S, Díaz-Rubio E, et al.: Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26 (12): 2013-9, 2008.
-
Heinemann V, von Weikersthal LF, Decker T, et al.: FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 15 (10): 1065-75, 2014.
-
Van Cutsem E, Tabernero J, Lakomy R, et al.: Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30 (28): 3499-506, 2012.
-
Grothey A, Van Cutsem E, Sobrero A, et al.: Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381 (9863): 303-12, 2013.
-
Gatalica Z, Torlakovic E: Pathology of the hereditary colorectal carcinoma. Fam Cancer 7 (1): 15-26, 2008.
-
O'Connell JB, Maggard MA, Livingston EH, et al.: Colorectal cancer in the young. Am J Surg 187 (3): 343-8, 2004.
-
Goel A, Nagasaka T, Spiegel J, et al.: Low frequency of Lynch syndrome among young patients with non-familial colorectal cancer. Clin Gastroenterol Hepatol 8 (11): 966-71, 2010.
-
Weber ML, Schneider DT, Offenmüller S, et al.: Pediatric Colorectal Carcinoma is Associated With Excellent Outcome in the Context of Cancer Predisposition Syndromes. Pediatr Blood Cancer 63 (4): 611-7, 2016.
-
Erdman SH: Pediatric adenomatous polyposis syndromes: an update. Curr Gastroenterol Rep 9 (3): 237-44, 2007.
-
Turcot J, Despres JP, St Pierre F: Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum 2: 465-8, 1959 Sep-Oct.
-
Vogelstein B, Fearon ER, Hamilton SR, et al.: Genetic alterations during colorectal-tumor development. N Engl J Med 319 (9): 525-32, 1988.
-
Lynch PM, Ayers GD, Hawk E, et al.: The safety and efficacy of celecoxib in children with familial adenomatous polyposis. Am J Gastroenterol 105 (6): 1437-43, 2010.
-
Pratt CB, Jane JA: Multiple colorectal carcinomas, polyposis coli, and neurofibromatosis, followed by multiple glioblastoma multiforme. J Natl Cancer Inst 83 (12): 880-1, 1991.
-
Heath JA, Reece JC, Buchanan DD, et al.: Childhood cancers in families with and without Lynch syndrome. Fam Cancer 14 (4): 545-51, 2015.
-
Modlin IM, Sandor A: An analysis of 8305 cases of carcinoid tumors. Cancer 79 (4): 813-29, 1997.
-
Deans GT, Spence RA: Neoplastic lesions of the appendix. Br J Surg 82 (3): 299-306, 1995.
-
Doede T, Foss HD, Waldschmidt J: Carcinoid tumors of the appendix in children--epidemiology, clinical aspects and procedure. Eur J Pediatr Surg 10 (6): 372-7, 2000.
-
Quaedvlieg PF, Visser O, Lamers CB, et al.: Epidemiology and survival in patients with carcinoid disease in The Netherlands. An epidemiological study with 2391 patients. Ann Oncol 12 (9): 1295-300, 2001.
-
Broaddus RR, Herzog CE, Hicks MJ: Neuroendocrine tumors (carcinoid and neuroendocrine carcinoma) presenting at extra-appendiceal sites in childhood and adolescence. Arch Pathol Lab Med 127 (9): 1200-3, 2003.
-
Foley DS, Sunil I, Debski R, et al.: Primary hepatic carcinoid tumor in children. J Pediatr Surg 43 (11): e25-8, 2008.
-
Tormey WP, FitzGerald RJ: The clinical and laboratory correlates of an increased urinary 5-hydroxyindoleacetic acid. Postgrad Med J 71 (839): 542-5, 1995.
-
Degnan AJ, Tocchio S, Kurtom W, et al.: Pediatric neuroendocrine carcinoid tumors: Management, pathology, and imaging findings in a pediatric referral center. Pediatr Blood Cancer : , 2017.
-
Delaunoit T, Rubin J, Neczyporenko F, et al.: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumors. Mayo Clin Proc 80 (4): 502-6, 2005.
-
More J, Young J, Reznik Y, et al.: Ectopic ACTH syndrome in children and adolescents. J Clin Endocrinol Metab 96 (5): 1213-22, 2011.
-
Pelizzo G, La Riccia A, Bouvier R, et al.: Carcinoid tumors of the appendix in children. Pediatr Surg Int 17 (5-6): 399-402, 2001.
-
Hatzipantelis E, Panagopoulou P, Sidi-Fragandrea V, et al.: Carcinoid tumors of the appendix in children: experience from a tertiary center in northern Greece. J Pediatr Gastroenterol Nutr 51 (5): 622-5, 2010.
-
Henderson L, Fehily C, Folaranmi S, et al.: Management and outcome of neuroendocrine tumours of the appendix-a two centre UK experience. J Pediatr Surg 49 (10): 1513-7, 2014.
-
Dall'Igna P, Ferrari A, Luzzatto C, et al.: Carcinoid tumor of the appendix in childhood: the experience of two Italian institutions. J Pediatr Gastroenterol Nutr 40 (2): 216-9, 2005.
-
Boxberger N, Redlich A, Böger C, et al.: Neuroendocrine tumors of the appendix in children and adolescents. Pediatr Blood Cancer 60 (1): 65-70, 2013.
-
Virgone C, Cecchetto G, Alaggio R, et al.: Appendiceal neuroendocrine tumours in childhood: Italian TREP project. J Pediatr Gastroenterol Nutr 58 (3): 333-8, 2014.
-
Boston CH, Phan A, Munsell MF, et al.: A Comparison Between Appendiceal and Nonappendiceal Neuroendocrine Tumors in Children and Young Adults: A Single-institution Experience. J Pediatr Hematol Oncol 37 (6): 438-42, 2015.
-
Corless CL, Fletcher JA, Heinrich MC: Biology of gastrointestinal stromal tumors. J Clin Oncol 22 (18): 3813-25, 2004.
-
Pappo AS, Janeway K, Laquaglia M, et al.: Special considerations in pediatric gastrointestinal tumors. J Surg Oncol 104 (8): 928-32, 2011.
-
Prakash S, Sarran L, Socci N, et al.: Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 27 (4): 179-87, 2005.
-
Miettinen M, Lasota J, Sobin LH: Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 29 (10): 1373-81, 2005.
-
Benesch M, Wardelmann E, Ferrari A, et al.: Gastrointestinal stromal tumors (GIST) in children and adolescents: A comprehensive review of the current literature. Pediatr Blood Cancer 53 (7): 1171-9, 2009.
-
Cypriano MS, Jenkins JJ, Pappo AS, et al.: Pediatric gastrointestinal stromal tumors and leiomyosarcoma. Cancer 101 (1): 39-50, 2004.
-
Pappo AS, Janeway KA: Pediatric gastrointestinal stromal tumors. Hematol Oncol Clin North Am 23 (1): 15-34, vii, 2009.
-
Benesch M, Leuschner I, Wardelmann E, et al.: Gastrointestinal stromal tumours in children and young adults: a clinicopathologic series with long-term follow-up from the database of the Cooperative Weichteilsarkom Studiengruppe (CWS). Eur J Cancer 47 (11): 1692-8, 2011.
-
Miettinen M, Lasota J: Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 130 (10): 1466-78, 2006.
-
Miettinen M, Lasota J: Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) - a review. Int J Biochem Cell Biol 53: 514-9, 2014.
-
Miettinen M, Wang ZF, Sarlomo-Rikala M, et al.: Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 35 (11): 1712-21, 2011.
-
Janeway KA, Liegl B, Harlow A, et al.: Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res 67 (19): 9084-8, 2007.
-
Tarn C, Rink L, Merkel E, et al.: Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proceedings of the National Academy of Sciences 105 (24): 8387-92, 2008. Also available online. Last accessed April 04, 2017.
-
Agaram NP, Laquaglia MP, Ustun B, et al.: Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res 14 (10): 3204-15, 2008.
-
Janeway KA, Kim SY, Lodish M, et al.: Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 108 (1): 314-8, 2011.
-
Killian JK, Miettinen M, Walker RL, et al.: Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med 6 (268): 268ra177, 2014.
-
Otto C, Agaimy A, Braun A, et al.: Multifocal gastric gastrointestinal stromal tumors (GISTs) with lymph node metastases in children and young adults: a comparative clinical and histomorphological study of three cases including a new case of Carney triad. Diagn Pathol 6: 52, 2011.
-
Carney JA: Carney triad: a syndrome featuring paraganglionic, adrenocortical, and possibly other endocrine tumors. J Clin Endocrinol Metab 94 (10): 3656-62, 2009.
-
Pasini B, McWhinney SR, Bei T, et al.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 16 (1): 79-88, 2008.
-
Demetri GD, Benjamin RS, Blanke CD, et al.: NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)--update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5 (Suppl 2): S1-29; quiz S30, 2007.
-
Janeway KA, Weldon CB: Pediatric gastrointestinal stromal tumor. Semin Pediatr Surg 21 (1): 31-43, 2012.
-
Demetri GD, van Oosterom AT, Garrett CR, et al.: Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368 (9544): 1329-38, 2006.
-
Demetri GD, von Mehren M, Blanke CD, et al.: Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347 (7): 472-80, 2002.
-
Janeway KA, Albritton KH, Van Den Abbeele AD, et al.: Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 52 (7): 767-71, 2009.
-
Dematteo RP, Ballman KV, Antonescu CR, et al.: Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 373 (9669): 1097-104, 2009.
Genital / Urinary TumorsGenital/urinary tumors include the following: - Carcinoma of the bladder.
- Testicular cancer (non-germ cell).
- Ovarian cancer (non-germ cell).
- Carcinoma of the cervix and vagina.
The prognosis, diagnosis, classification, and treatment of these genital/urinary tumors are discussed below. It must be emphasized that these tumors are seen very infrequently in patients younger than 15 years, and most of the evidence is derived from case series. Carcinoma of the Bladder Urothelial bladder neoplasms are extremely rare in children. Histologic classification of these neoplasms includes urothelial papillomas, papillary neoplasms of low malignant potential, low-grade urothelial carcinoma, and high-grade urothelial carcinoma. An alternative designation is transitional cell carcinoma of the bladder. The most common histology is papillary urothelial neoplasm of low malignant potential, while high-grade, invasive urothelial carcinomas are extremely rare in young patients.[1,2,3,4,5] Bladder cancer in adolescents may develop as a consequence of alkylating-agent chemotherapy given for other childhood tumors or leukemia.[4,6,7] The association between cyclophosphamide and bladder cancer is the only established relationship between a specific anticancer drug and a solid tumor.[6] Treatment and Outcome In contrast to adults, most pediatric bladder carcinomas are low grade, superficial, and have an excellent prognosis after transurethral resection.[2,3,5,8] Squamous cell carcinoma and more aggressive carcinomas, however, have been reported and may require a more aggressive surgical approach.[3,9,10,11] (Refer to the PDQ summary on adult Bladder Cancer Treatment for more information.) Testicular Cancer (Non-Germ Cell) Testicular tumors are very rare in young boys and account for an incidence of 1% to 2% of all childhood tumors.[12,13] The most common testicular tumors are benign teratomas followed by malignant nonseminomatous germ cell tumors. (Refer to the PDQ summary on Childhood Extracranial Germ Cell Tumors for more information.) Non-germ cell tumors such as sex cord-stromal tumors are exceedingly rare in prepubertal boys. In a small series, gonadal stromal tumors accounted for 8% to 13% of pediatric testicular tumors.[14,15] In newborns and infants, juvenile granulosa cell and Sertoli cell tumors are the most common stromal cell tumor.[16] Juvenile granulosa cell tumors usually present in infancy (median age, 6 days) and Sertoli cell tumors present later in infancy (median age, 7 months). In older males, Leydig cell tumors are more common. The prognosis for sex cord-stromal tumors is usually excellent after orchiectomy.[17,18,19]; [20][Level of evidence: 3iiiA] In a review of the literature, 79 patients younger than 12 years were identified. No patient had high-risk pathological findings after orchiectomy, and none had evidence of occult metastatic disease, suggesting a role for a limited surveillance strategy.[21][Level of evidence: 3iiiA] Treatment There are conflicting data about malignant potential in older males. Most case reports suggest that in the pediatric patients, these tumors can be treated with surgery alone.[17][Level of evidence: 3iii]; [22][Level of evidence: 3iiiA]; [19][Level of evidence: 3iiiDii] It is prudent to check alpha-fetoprotein (AFP) levels before surgery. Elevated AFP levels are usually indicative of a malignant germ cell tumor. However, AFP levels and decay in levels are often difficult to interpret in infants younger than 1 year.[23] - In a retrospective study, 42 patients with sex cord-stromal tumors were identified. All tumors were confined to the testes. They were treated with surgery alone, according to specific germ cell tumor guidelines. There were no recurrences.[20][Level of evidence: 3iiiA]
- A French registry identified 11 boys with localized sex cord-stromal testicular tumors.[24][Level of evidence: 3iA] All 11 boys were treated with surgery alone; none had a recurrence. The benign behavior of pediatric non-germ cell testicular tumors has led to reports of testis-sparing surgery.[25,26]
However, given the rarity of this tumor, the surgical approach in pediatrics has not been well defined. Ovarian Cancer (Non-Germ Cell) The majority of ovarian masses in children are not malignant. The most common neoplasms are germ cell tumors, followed by epithelial tumors, stromal tumors, and then other tumors such as Burkitt lymphoma.[27,28,29,30] The majority of malignant ovarian tumors occur in girls aged 15 to 19 years.[31] Epithelial Ovarian Neoplasia Ovarian tumors derived from malignant epithelial elements include adenocarcinomas, cystadenocarcinomas, (mucinous) borderline tumors, endometrioid tumors, clear cell tumors, and undifferentiated carcinomas.[32] In one series of 19 patients younger than 21 years with epithelial ovarian neoplasms, the average age at diagnosis was 19.7 years. Dysmenorrhea and abdominal pain were the most common presenting symptoms; 79% of the patients had stage I disease with a 100% survival rate, and only those who had small cell anaplastic carcinoma died.[33] Girls with ovarian carcinoma (epithelial ovarian neoplasia) fare better than do adults with similar histology, probably because girls usually present with low-stage disease.[33] Treatment is stage-related and may include surgery, radiation, and chemotherapy with cisplatin, carboplatin, etoposide, topotecan, paclitaxel, and other agents. (Refer to the PDQ summary on adult Ovarian Epithelial, Fallopian Tube, and Primary Peritoneal Cancer Treatment for more information.) Ovarian surface epithelial neoplasms comprise a small subset of ovarian epithelial neoplasms; in children, most of the cases are of serous or mucinous histology and have a low malignant potential. Surgery and chemotherapy have been used to treat ovarian surface epithelial neoplasms.[34] Sex Cord-Stromal Tumors Ovarian sex cord-stromal tumors are a heterogeneous group of rare tumors that derive from the gonadal non-germ cell component.[35] Histologic subtypes display some areas of gonadal differentiation and include juvenile granulosa cell tumors, Sertoli-Leydig cell tumors, and sclerosing stromal tumors. Ovarian Sertoli-Leydig cell tumors in children and adolescents are commonly associated with the presence of germline DICER1 mutations and may be a manifestation of the familial pleuropulmonary blastoma syndrome.[36] The clinical presentation and prognosis of sex cord-stromal tumors varies by histology. In all entities, metastatic spread occurs rarely and if present, is usually limited to the peritoneal cavity.[35] Distant metastases may rarely occur, mostly in relapse situations.[37] In the United States, these tumors may be registered in the Testicular and Ovarian Stromal Tumor registry.[38] In Europe, patients are prospectively registered in the national rare tumor groups.[38,39] The recommendations regarding diagnostic work-up, staging, and therapeutic strategy have been harmonized between these registries.[38] A French registry identified 38 girls younger than 18 years with ovarian sex cord tumors.[24] Complete surgical resection was achieved in 23 of 38 girls who did not receive adjuvant treatment. Two patients recurred, one patient's tumor responded to chemotherapy, and the other patient died. Fifteen girls had tumor rupture and/or ascites. Eleven of the 15 patients received chemotherapy and did not recur; of the four who did not receive chemotherapy, all recurred and two died. Juvenile Granulosa Cell Tumors The most common histologic subtype in girls younger than 18 years is juvenile granulosa cell tumors (median age, 7.6 years; range, birth to 17.5 years).[40,41] Juvenile granulosa cell tumors represent about 5% of ovarian tumors in children and adolescents and are distinct from the granulosa cell tumors seen in adults.[35,42,43,44] Patients with juvenile granulosa cell tumors present with the following:[45,46] - Precocious puberty (most common).
- Abdominal pain.
- Abdominal mass.
- Ascites.
Juvenile granulosa cell tumors have been reported in children with Ollier disease and Maffucci syndrome. As many as 90% of children with juvenile granulosa cell tumors will have low-stage disease (stage I) by International Federation of Gynecology and Obstetrics (FIGO) criteria and are usually curable with unilateral salpingo-oophorectomy alone. Patients with spontaneous tumor rupture or malignant ascites (FIGO stage Ic), advanced disease (FIGO stage II-IV), and those with high mitotic activity tumors have a poorer prognosis and require chemotherapy.[24,39] Use of a cisplatin-based chemotherapy regimen has been reported in both the adjuvant and recurrent disease settings with some success.[39,40,44,47,48] Sertoli-Leydig Cell Tumors Sertoli-Leydig cell tumors are rare in young girls and are more frequently seen in adolescents. They may present with virilization [49] or precocious puberty.[50] These tumors may also be associated with Peutz-Jeghers syndrome, but more frequently are a part of the DICER-1 tumor spectrum.[36,51,52] A study of 44 patients from the European Cooperative Study Group on Pediatric Rare Tumors showed that prognosis of Sertoli-Leydig tumors was determined by stage and histopathologic differentiation.[53] Surgery is the primary treatment for Sertoli-Leydig cell tumors and is the only treatment for low-stage disease (FIGO stage Ia), with essentially 100% event-free survival.[24] Patients with Sertoli-Leydig tumors with abdominal spillage during surgery, spontaneous tumor rupture, or metastatic disease (FIGO stages IC, II, III, and IV) are treated with cisplatin-based combination chemotherapy, although the impact of chemotherapy has not been studied in clinical trials.[24,53] An additional study reported on 40 women with FIGO stage I or Ic Sertoli-Leydig cell tumors of the ovary, with an average age of 28 years.[54][Level of evidence: 3iiA] Of 34 patients with intermediate or poor differentiation, 23 patients received postoperative chemotherapy (most regimens included cisplatin); none recurred. Of the 11 patients who did not receive postoperative chemotherapy, two recurred; both had tumors that were salvaged with chemotherapy. Small Cell Carcinoma of the Ovary Small cell carcinomas of the ovary are exceedingly rare and aggressive tumors and may be associated with hypercalcemia.[55] Successful treatment with aggressive therapy has been reported in a few cases.[55,56][Level of evidence: 3iiB]; [57,58][Level of evidence: 3iiiA] Carcinoma of the Cervix and Vagina Incidence, Risk Factors, and Clinical Presentation Adenocarcinoma of the cervix and vagina is rare in childhood and adolescence, with fewer than 50 reported cases.[30,59] Two-thirds of the cases are related to exposure to diethylstilbestrol in utero. The median age at presentation is 15 years, with a range of 7 months to 18 years, and most patients present with vaginal bleeding. Adults with adenocarcinoma of the cervix or vagina will present with stage I or stage II disease 90% of the time. In children and adolescents, there is a high incidence of stage III and stage IV disease (24%). This difference may be explained by the practice of routine pelvic examinations in adults and the hesitancy to perform pelvic exams in children. Treatment and Outcome The treatment of choice is surgical resection,[60] followed by radiation therapy for residual microscopic disease or lymphatic metastases. The role of chemotherapy in management is unknown, although drugs commonly used in the treatment of gynecologic malignancies, carboplatin and paclitaxel, have been used. The 3-year event-free survival (EFS) for all stages is 71% ± 11%; for stage I and stage II, the EFS is 82% ± 11%, and for stage III and stage IV, the EFS is 57% ± 22%.[59] References:
-
Alanee S, Shukla AR: Bladder malignancies in children aged <18 years: results from the Surveillance, Epidemiology and End Results database. BJU Int 106 (4): 557-60, 2010.
-
Paner GP, Zehnder P, Amin AM, et al.: Urothelial neoplasms of the urinary bladder occurring in young adult and pediatric patients: a comprehensive review of literature with implications for patient management. Adv Anat Pathol 18 (1): 79-89, 2011.
-
Stanton ML, Xiao L, Czerniak BA, et al.: Urothelial tumors of the urinary bladder in young patients: a clinicopathologic study of 59 cases. Arch Pathol Lab Med 137 (10): 1337-41, 2013.
-
Di Carlo D, Ferrari A, Perruccio K, et al.: Management and follow-up of urothelial neoplasms of the bladder in children: a report from the TREP project. Pediatr Blood Cancer 62 (6): 1000-3, 2015.
-
Berrettini A, Castagnetti M, Salerno A, et al.: Bladder urothelial neoplasms in pediatric age: experience at three tertiary centers. J Pediatr Urol 11 (1): 26.e1-5, 2015.
-
Johansson SL, Cohen SM: Epidemiology and etiology of bladder cancer. Semin Surg Oncol 13 (5): 291-8, 1997 Sep-Oct.
-
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. International Agency for Research on Cancer: Overall evaluations of carcinogenicity: an updating of IARC monographs, volumes 1 to 42. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Supplement 7. Lyon, France: International Agency for Research on Cancer, 1987.
-
Fine SW, Humphrey PA, Dehner LP, et al.: Urothelial neoplasms in patients 20 years or younger: a clinicopathological analysis using the world health organization 2004 bladder consensus classification. J Urol 174 (5): 1976-80, 2005.
-
Sung JD, Koyle MA: Squamous cell carcinoma of the bladder in a pediatric patient. J Pediatr Surg 35 (12): 1838-9, 2000.
-
Lezama-del Valle P, Jerkins GR, Rao BN, et al.: Aggressive bladder carcinoma in a child. Pediatr Blood Cancer 43 (3): 285-8, 2004.
-
Korrect GS, Minevich EA, Sivan B: High-grade transitional cell carcinoma of the pediatric bladder. J Pediatr Urol 8 (3): e36-8, 2012.
-
Hartke DM, Agarwal PK, Palmer JS: Testicular neoplasms in the prepubertal male. J Mens Health Gend 3 (2): 131-8, 2006.
-
Ahmed HU, Arya M, Muneer A, et al.: Testicular and paratesticular tumours in the prepubertal population. Lancet Oncol 11 (5): 476-83, 2010.
-
Pohl HG, Shukla AR, Metcalf PD, et al.: Prepubertal testis tumors: actual prevalence rate of histological types. J Urol 172 (6 Pt 1): 2370-2, 2004.
-
Schwentner C, Oswald J, Rogatsch H, et al.: Stromal testis tumors in infants. a report of two cases. Urology 62 (6): 1121, 2003.
-
Carmignani L, Colombo R, Gadda F, et al.: Conservative surgical therapy for leydig cell tumor. J Urol 178 (2): 507-11; discussion 511, 2007.
-
Agarwal PK, Palmer JS: Testicular and paratesticular neoplasms in prepubertal males. J Urol 176 (3): 875-81, 2006.
-
Dudani R, Giordano L, Sultania P, et al.: Juvenile granulosa cell tumor of testis: case report and review of literature. Am J Perinatol 25 (4): 229-31, 2008.
-
Cecchetto G, Alaggio R, Bisogno G, et al.: Sex cord-stromal tumors of the testis in children. A clinicopathologic report from the Italian TREP project. J Pediatr Surg 45 (9): 1868-73, 2010.
-
Hofmann M, Schlegel PG, Hippert F, et al.: Testicular sex cord stromal tumors: analysis of patients from the MAKEI study. Pediatr Blood Cancer 60 (10): 1651-5, 2013.
-
Rove KO, Maroni PD, Cost CR, et al.: Pathologic Risk Factors in Pediatric and Adolescent Patients With Clinical Stage I Testicular Stromal Tumors. J Pediatr Hematol Oncol 37 (8): e441-6, 2015.
-
Thomas JC, Ross JH, Kay R: Stromal testis tumors in children: a report from the prepubertal testis tumor registry. J Urol 166 (6): 2338-40, 2001.
-
Blohm ME, Vesterling-Hörner D, Calaminus G, et al.: Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 15 (2): 135-42, 1998 Mar-Apr.
-
Fresneau B, Orbach D, Faure-Conter C, et al.: Sex-Cord Stromal Tumors in Children and Teenagers: Results of the TGM-95 Study. Pediatr Blood Cancer 62 (12): 2114-9, 2015.
-
Cosentino M, Algaba F, Saldaña L, et al.: Juvenile granulosa cell tumor of the testis: a bilateral and synchronous case. Should testis-sparing surgery be mandatory? Urology 84 (3): 694-6, 2014.
-
Kao CS, Cornejo KM, Ulbright TM, et al.: Juvenile granulosa cell tumors of the testis: a clinicopathologic study of 70 cases with emphasis on its wide morphologic spectrum. Am J Surg Pathol 39 (9): 1159-69, 2015.
-
Morowitz M, Huff D, von Allmen D: Epithelial ovarian tumors in children: a retrospective analysis. J Pediatr Surg 38 (3): 331-5; discussion 331-5, 2003.
-
Schultz KA, Sencer SF, Messinger Y, et al.: Pediatric ovarian tumors: a review of 67 cases. Pediatr Blood Cancer 44 (2): 167-73, 2005.
-
Aggarwal A, Lucco KL, Lacy J, et al.: Ovarian epithelial tumors of low malignant potential: a case series of 5 adolescent patients. J Pediatr Surg 44 (10): 2023-7, 2009.
-
You W, Dainty LA, Rose GS, et al.: Gynecologic malignancies in women aged less than 25 years. Obstet Gynecol 105 (6): 1405-9, 2005.
-
Brookfield KF, Cheung MC, Koniaris LG, et al.: A population-based analysis of 1037 malignant ovarian tumors in the pediatric population. J Surg Res 156 (1): 45-9, 2009.
-
Lovvorn HN 3rd, Tucci LA, Stafford PW: Ovarian masses in the pediatric patient. AORN J 67 (3): 568-76; quiz 577, 580-84, 1998.
-
Tsai JY, Saigo PE, Brown C, et al.: Diagnosis, pathology, staging, treatment, and outcome of epithelial ovarian neoplasia in patients age < 21 years. Cancer 91 (11): 2065-70, 2001.
-
Hazard FK, Longacre TA: Ovarian surface epithelial neoplasms in the pediatric population: incidence, histologic subtype, and natural history. Am J Surg Pathol 37 (4): 548-53, 2013.
-
Schneider DT, Jänig U, Calaminus G, et al.: Ovarian sex cord-stromal tumors--a clinicopathological study of 72 cases from the Kiel Pediatric Tumor Registry. Virchows Arch 443 (4): 549-60, 2003.
-
Schultz KA, Pacheco MC, Yang J, et al.: Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 122 (2): 246-50, 2011.
-
Wessalowski R, Spaar HJ, Pape H, et al.: Successful liver treatment of a juvenile granulosa cell tumor in a 4-year-old child by regional deep hyperthermia, systemic chemotherapy, and irradiation. Gynecol Oncol 57 (3): 417-22, 1995.
-
Schultz KA, Schneider DT, Pashankar F, et al.: Management of ovarian and testicular sex cord-stromal tumors in children and adolescents. J Pediatr Hematol Oncol 34 (Suppl 2): S55-63, 2012.
-
Schneider DT, Calaminus G, Harms D, et al.: Ovarian sex cord-stromal tumors in children and adolescents. J Reprod Med 50 (6): 439-46, 2005.
-
Calaminus G, Wessalowski R, Harms D, et al.: Juvenile granulosa cell tumors of the ovary in children and adolescents: results from 33 patients registered in a prospective cooperative study. Gynecol Oncol 65 (3): 447-52, 1997.
-
Capito C, Flechtner I, Thibaud E, et al.: Neonatal bilateral ovarian sex cord stromal tumors. Pediatr Blood Cancer 52 (3): 401-3, 2009.
-
Bouffet E, Basset T, Chetail N, et al.: Juvenile granulosa cell tumor of the ovary in infants: a clinicopathologic study of three cases and review of the literature. J Pediatr Surg 32 (5): 762-5, 1997.
-
Zaloudek C, Norris HJ: Granulosa tumors of the ovary in children: a clinical and pathologic study of 32 cases. Am J Surg Pathol 6 (6): 503-12, 1982.
-
Vassal G, Flamant F, Caillaud JM, et al.: Juvenile granulosa cell tumor of the ovary in children: a clinical study of 15 cases. J Clin Oncol 6 (6): 990-5, 1988.
-
Kalfa N, Patte C, Orbach D, et al.: A nationwide study of granulosa cell tumors in pre- and postpubertal girls: missed diagnosis of endocrine manifestations worsens prognosis. J Pediatr Endocrinol Metab 18 (1): 25-31, 2005.
-
Gell JS, Stannard MW, Ramnani DM, et al.: Juvenile granulosa cell tumor in a 13-year-old girl with enchondromatosis (Ollier's disease): a case report. J Pediatr Adolesc Gynecol 11 (3): 147-50, 1998.
-
Powell JL, Connor GP, Henderson GS: Management of recurrent juvenile granulosa cell tumor of the ovary. Gynecol Oncol 81 (1): 113-6, 2001.
-
Schneider DT, Calaminus G, Wessalowski R, et al.: Therapy of advanced ovarian juvenile granulosa cell tumors. Klin Padiatr 214 (4): 173-8, 2002 Jul-Aug.
-
Arhan E, Cetinkaya E, Aycan Z, et al.: A very rare cause of virilization in childhood: ovarian Leydig cell tumor. J Pediatr Endocrinol Metab 21 (2): 181-3, 2008.
-
Choong CS, Fuller PJ, Chu S, et al.: Sertoli-Leydig cell tumor of the ovary, a rare cause of precocious puberty in a 12-month-old infant. J Clin Endocrinol Metab 87 (1): 49-56, 2002.
-
Zung A, Shoham Z, Open M, et al.: Sertoli cell tumor causing precocious puberty in a girl with Peutz-Jeghers syndrome. Gynecol Oncol 70 (3): 421-4, 1998.
-
Schultz KA, Harris A, Messinger Y, et al.: Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer 15 (1): 105-10, 2016.
-
Schneider DT, Orbach D, Cecchetto G, et al.: Ovarian Sertoli Leydig cell tumours in children and adolescents: an analysis of the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Eur J Cancer 51 (4): 543-50, 2015.
-
Gui T, Cao D, Shen K, et al.: A clinicopathological analysis of 40 cases of ovarian Sertoli-Leydig cell tumors. Gynecol Oncol 127 (2): 384-9, 2012.
-
Distelmaier F, Calaminus G, Harms D, et al.: Ovarian small cell carcinoma of the hypercalcemic type in children and adolescents: a prognostically unfavorable but curable disease. Cancer 107 (9): 2298-306, 2006.
-
Pressey JG, Kelly DR, Hawthorne HT: Successful treatment of preadolescents with small cell carcinoma of the ovary hypercalcemic type. J Pediatr Hematol Oncol 35 (7): 566-9, 2013.
-
Christin A, Lhomme C, Valteau-Couanet D, et al.: Successful treatment for advanced small cell carcinoma of the ovary. Pediatr Blood Cancer 50 (6): 1276-7, 2008.
-
Kanwar VS, Heath J, Krasner CN, et al.: Advanced small cell carcinoma of the ovary in a seventeen-year-old female, successfully treated with surgery and multi-agent chemotherapy. Pediatr Blood Cancer 50 (5): 1060-2, 2008.
-
McNall RY, Nowicki PD, Miller B, et al.: Adenocarcinoma of the cervix and vagina in pediatric patients. Pediatr Blood Cancer 43 (3): 289-94, 2004.
-
Abu-Rustum NR, Su W, Levine DA, et al.: Pediatric radical abdominal trachelectomy for cervical clear cell carcinoma: a novel surgical approach. Gynecol Oncol 97 (1): 296-300, 2005.
Other Rare Childhood CancersOther rare childhood cancers include the following: - Multiple endocrine neoplasia (MEN) syndromes and Carney complex.
- Pheochromocytoma and paraganglioma.
- Skin cancer (melanoma, basal cell carcinoma, and squamous cell carcinoma).
- Intraocular (Uveal) Melanoma.
- Chordoma.
- Cancer of unknown primary site.
The prognosis, diagnosis, classification, and treatment of these other rare childhood cancers are discussed below. It must be emphasized that these cancers are seen very infrequently in patients younger than 15 years, and most of the evidence is derived from case series. Multiple Endocrine Neoplasia (MEN) Syndromes and Carney Complex MEN syndromes are familial disorders characterized by neoplastic changes that affect multiple endocrine organs.[1] Changes may include hyperplasia, benign adenomas, and carcinomas. There are two main types of MEN syndrome: - Type 1.
- Type 2.
- Type 2A.
- Type 2B.
- Familial medullary thyroid carcinoma.
(Refer to the PDQ summary on Genetics of Endocrine and Neuroendocrine Neoplasias for more information about MEN syndromes.) Clinical Presentation and Diagnostic Evaluation The most salient clinical and genetic alterations of the multiple endocrine neoplasia (MEN) syndromes are shown in Table 7. Table 7. Multiple Endocrine Neoplasia (MEN) Syndromes with Associated Clinical and Genetic Alterations| Syndrome | Clinical Features/Tumors | Genetic Alterations |
|---|
| MEN type 1: Werner syndrome[2] | Parathyroid | 11q13 (MEN1 gene) | | Pancreatic islets: | Gastrinoma | 11q13 (MEN1 gene) | | Insulinoma | | Glucagonoma | | VIPoma | | Pituitary: | Prolactinoma | 11q13 (MEN1 gene) | | Somatotrophinoma | | Corticotropinoma | | Other associated tumors (less common): | Carcinoid: bronchial and thymic | 11q13 (MEN1 gene) | | Adrenocortical | | Lipoma | | Angiofibroma | | Collagenoma | | MEN type 2A: Sipple syndrome | Medullary thyroid carcinoma | 10q11.2 (RET gene) | | Pheochromocytoma | | Parathyroid gland | | MEN type 2B | Medullary thyroid carcinoma | 10q11.2 (RET gene) | | Pheochromocytoma | | Mucosal neuromas | | Intestinal ganglioneuromatosis | | Marfanoid habitus | - Multiple endocrine neoplasia type 1 (MEN1) syndrome (Werner syndrome): MEN1 syndrome is an autosomal dominant disorder characterized by the presence of tumors in the parathyroid, pancreatic islet cells, and anterior pituitary. Diagnosis of this syndrome should be considered when two endocrine tumors listed in Table 7 are present.
A study documented the initial symptoms of MEN1 syndrome occurring before age 21 years in 160 patients.[3] Of note, most patients had familial MEN1 syndrome and were followed up using an international screening protocol. - Primary hyperparathyroidism. Primary hyperparathyroidism, the most common symptom, was found in 75% of patients, usually only in those with biological abnormalities. Primary hyperparathyroidism diagnosed outside of a screening program is extremely rare, most often presents with nephrolithiasis, and should lead the clinician to suspect MEN1.[3,4]
- Pituitary adenomas. Pituitary adenomas were discovered in 34% of patients, occurred mainly in females older than 10 years, and were often symptomatic.[3]
- Pancreatic neuroendocrine tumors. Pancreatic neuroendocrine tumors were found in 23% of patients. Specific diagnoses included insulinoma, nonsecreting pancreatic tumor, and Zollinger-Ellison syndrome. The first case of insulinoma occurred before age 5 years.[3]
- Malignant tumors. Four patients had malignant tumors (two adrenal carcinomas, one gastrinoma, and one thymic carcinoma). The patient with thymic carcinoma died before age 21 years from rapidly progressive disease.
Germline mutations of the MEN1 gene located on chromosome 11q13 are found in 70% to 90% of patients; however, this gene has also been shown to be frequently inactivated in sporadic tumors.[5] Mutation testing is combined with clinical screening for patients and family members with proven at-risk MEN1 syndrome.[6] It is recommended that screening for patients with MEN1 syndrome begin by the age of 5 years and continue for life. The number of tests or biochemical screening is age specific and may include yearly serum calcium, parathyroid hormone, gastrin, glucagon, secretin, proinsulin, chromogranin A, prolactin, and IGF-1. Radiologic screening should include a magnetic resonance imaging of the brain and computed tomography of the abdomen every 1 to 3 years.[7] - Multiple endocrine neoplasia type 2A (MEN2A) and multiple endocrine neoplasia type 2B (MEN2B) syndromes:
A germline activating mutation in the RET oncogene (a receptor tyrosine kinase) on chromosome 10q11.2 is responsible for the uncontrolled growth of cells in medullary thyroid carcinoma associated with MEN2A and MEN2B syndromes.[8,9,10] - MEN2A: MEN2A is characterized by the presence of two or more endocrine tumors (refer to Table 7) in an individual or in close relatives.[11]RET mutations in these patients are usually confined to exons 10 and 11.
- MEN2B: MEN2B is characterized by medullary thyroid carcinomas, parathyroid hyperplasias, adenomas, pheochromocytomas, mucosal neuromas, and ganglioneuromas.[11,12,13] The medullary thyroid carcinomas that develop in these patients are extremely aggressive. More than 95% of mutations in these patients are confined to codon 918 in exon 16, causing receptor autophosphorylation and activation.[14] Patients also have medullated corneal nerve fibers, distinctive faces with enlarged lips, and an asthenic Marfanoid body habitus.
A pentagastrin stimulation test can be used to detect the presence of medullary thyroid carcinoma in these patients, although management of patients is driven primarily by the results of genetic analysis for RET mutations.[14,15]
Guidelines for genetic testing of suspected patients with MEN2 syndrome and the correlations between the type of mutation and the risk levels of aggressiveness of medullary thyroid cancer have been published.[15,16] - Familial Medullary Thyroid Carcinoma: Familial medullary thyroid carcinoma is diagnosed in families with medullary thyroid carcinoma in the absence of pheochromocytoma or parathyroid adenoma/hyperplasia. RET mutations in exons 10, 11, 13, and 14 account for most cases.
The most-recent literature suggests that this entity should not be identified as a form of hereditary medullary thyroid carcinoma that is separate from MEN2A and MEN2B. Familial medullary thyroid carcinoma should be recognized as a variant of MEN2A, to include families with only medullary thyroid cancer who meet the original criteria for familial disease. The original criteria includes families of at least two generations with at least two, but less than ten, patients with RET germline mutations; small families in which two or fewer members in a single generation have germline RET mutations; and single individuals with a RET germline mutation.[15,17]
Table 8. Clinical Features of Multiple Endocrine Neoplasia Type 2 (MEN2) Syndromes| MEN2 Subtype | Medullary Thyroid Carcinoma | Pheochromocytoma | Parathyroid Disease |
|---|
| MEN2A | 95% | 50% | 20% to 30% | | MEN2B | 100% | 50% | Uncommon | Treatment - MEN1 syndrome: Treatment of patients with MEN1 syndrome is based on the type of tumor. The outcome of patients with MEN1 syndrome is generally good provided adequate treatment can be obtained for parathyroid, pancreatic, and pituitary tumors.
The standard approach to patients who present with hyperparathyroidism and MEN1 syndrome is genetic testing and treatment with a cervical resection of at least three parathyroid glands and transcervical thymectomy.[4] - MEN2 syndromes: The management of medullary thyroid cancer in children from families having MEN2 syndromes relies on presymptomatic detection of the RET proto-oncogene mutation responsible for the disease.
- MEN2A syndrome: For children with MEN2A, thyroidectomy is commonly performed by approximately age 5 years or older if that is when a mutation is identified. [10,18,19,20,21,22] The outcome for patients with MEN2A syndrome is also generally good, yet the possibility exists for recurrence of medullary thyroid carcinoma and pheochromocytoma.[23,24,25]
Relatives of patients with MEN2A undergo genetic testing in early childhood, before the age of 5 years. Carriers undergo total thyroidectomy as described above with autotransplantation of one parathyroid gland by a certain age.[22,26,27,28] - MEN2B syndrome: Because of the increased virulence of medullary thyroid carcinoma in children with MEN2B and in those with mutations in codons 883, 918, and 922, it is recommended that these children undergo prophylactic thyroidectomy in infancy.[14,19,29]; [30][Level of evidence: 3iiiDii] Patients who have MEN2B syndrome have a worse outcome primarily because of more aggressive medullary thyroid carcinoma. Prophylactic thyroidectomy has the potential to improve the outcome in MEN2B.[31]
Complete removal of the thyroid gland is the recommended procedure for surgical management of medullary thyroid cancer in children because there is a high incidence of bilateral disease. Hirschsprung disease has been associated in a small percentage of cases with the development of neuroendocrine tumors such as medullary thyroid carcinoma. RET germline inactivating mutations have been detected in up to 50% of patients with familial Hirschsprung disease and less often in the sporadic form.[32,33,34] Cosegregation of Hirschsprung disease and medullary thyroid carcinoma phenotype is infrequently reported, but these individuals usually have a mutation in RET exon 10. It has been recommended that patients with Hirschsprung disease be screened for mutations in RET exon 10 and consideration be given to prophylactic thyroidectomy if such a mutation is discovered.[34,35,36] (Refer to the PDQ summary on Genetics of Endocrine and Neuroendocrine Neoplasias for more information about MEN2A and MEN2B.)
In a randomized phase III trial for adult patients with unresectable locally advanced or metastatic hereditary or sporadic medullary thyroid carcinoma treated with either vandetanib (a selective inhibitor of RET, vascular endothelial growth factor receptor, and epidermal growth factor receptor) or placebo, vandetanib administration was associated with significant improvements in progression-free survival, response rate, disease control rates, and biochemical response.[37] Children with locally advanced or metastatic medullary thyroid carcinoma were treated with vandetanib in a phase I/II trial. Of 16 patients, only one had no response and seven had a partial response. Disease in three of those patients subsequently recurred, but 11 of 16 patients treated with vandetanib remained on therapy at the time of the report.[38] Carney Complex Carney complex is an autosomal dominant syndrome caused by mutations in the PPKAR1A gene, located in chromosome 17.[39] The syndrome is characterized by cardiac and cutaneous myxomas, pale brown to brown lentigines, blue nevi, primary pigmented nodular adrenocortical disease causing Cushing syndrome, and a variety of endocrine and nonendocrine tumors, including pituitary adenomas, thyroid tumors, and large cell calcifying Sertoli cell tumor of the testis.[39,40,41] There are published surveillance guidelines for patients with Carney complex that include cardiac, testicular, and thyroid ultrasound. For patients with the Carney complex, prognosis depends on the frequency of recurrences of cardiac and skin myxomas and other tumors. Pheochromocytoma and Paraganglioma Pheochromocytoma and paraganglioma are rare catecholamine-producing tumors with a combined annual incidence of three cases per 1 million individuals. Paraganglioma and pheochromocytoma are exceedingly rare in the pediatric and adolescent population, accounting for approximately 20% of all cases.[42,43] Tumors arising within the adrenal gland are known as pheochromocytomas, whereas morphologically identical tumors arising elsewhere are termed paragangliomas. Paragangliomas are further divided into the following subtypes:[44,45] - Sympathetic paragangliomas that predominantly arise from the intra-abdominal sympathetic trunk and usually produce catecholamines.
- Parasympathetic paragangliomas that are distributed along the parasympathetic nerves of the head, neck, and mediastinum and are rarely functional.
Risk Factors It is now estimated that up to 30% of all pheochromocytomas and paragangliomas are familial; several susceptibility genes have been described (refer to Table 9). The median age at presentation in most familial syndromes is 30 to 35 years, and up to 50% of subjects have disease by age 26 years.[46,47,48,49] Table 9. Characteristics of Paraganglioma (PGL) and Pheochromocytoma (PCC) Associated with Susceptibility Genesa| Germline Mutation | Syndrome | Proportion of all PGL/PCC (%) | Mean Age at Presentation (y) | Penetrance of PGL/PCC (%) |
|---|
| MEN1 = multiple endocrine neoplasia type 1; MEN2 = multiple endocrine neoplasia type 2; NF1 = neurofibromatosis type 1; VHL = von Hippel-Lindau. | | a Adapted from Welander et al.[46] | | RET | MEN2 | 5.3 | 35.6 | 50 | | VHL | VHL | 9.0 | 28.6 | 10-26 | | NF1 | NF1 | 2.9 | 41.6 | 0.1-5.7 | | SDHD | PGL1 | 7.1 | 35.0 | 86 | | SDHFA2 | PGL2 | <1 | 32.2 | 100 | | SDHC | PGL3 | <1 | 42.7 | Unknown | | SDHB | PGL4 | 5.5 | 32.7 | 77 | | SDHA | - | <3 | 40.0 | Unknown | | KIF1B-beta | - | <1 | 46.0 | Unknown | | EGLN1 | - | <1 | 43.0 | Unknown | | TMEM127 | - | <2 | 42.8 | Unknown | | MAX[49] | - | <2 | 34 | Unknown | | Unknown | Carney triad | <1 | 27.5 | - | | SDHB, C, D | Carney-Stratakis | <1 | 33 | Unknown | | MEN1 | MEN1 | <1 | 30.5 | Unknown | | No mutation | Sporadic disease | 70 | 48.3 | - | Genetic factors and syndromes associated with an increased risk of pheochromocytoma and paraganglioma include the following: - von Hippel-Lindau (VHL) syndrome: Pheochromocytoma and paraganglioma occur in 10% to 20% of patients with VHL.
- Multiple Endocrine Neoplasia (MEN) Syndrome Type 2: Codon-specific mutations of the RET gene are associated with a 50% risk of development of pheochromocytoma in MEN2A and MEN2B. Somatic RET mutations are also found in sporadic pheochromocytoma and paraganglioma.
- Neurofibromatosis type 1 (NF1): Pheochromocytoma and paraganglioma are a rare occurrence in patients with NF1, and typically have characteristics similar to those of sporadic tumors, with a relatively late mean age of onset and rarity in pediatrics.
- Familial pheochromocytoma/paraganglioma syndromes, associated with germline mutations of mitochondrial succinate dehydrogenase (SDH) complex genes (refer to Table 9). They are all inherited in an autosomal dominant manner but with varying penetrance.
- PGL1: Associated with SDHD mutations, manifests more commonly with head and neck paragangliomas, and has a very high penetrance, with more than 80% of carriers developing disease by age 50 years.
- PGL2: Associated with SDHAF2 mutations, is very rare, and generally manifests as parasympathetic paraganglioma.
- PGL3: Associated with SDHC mutations, is very rare, and usually presents with parasympathetic paraganglioma, often unifocal, benign, and in the head and neck location.
- PGL4: Associated with SDHB mutations and usually manifests with intra-abdominal sympathetic paraganglioma. The neoplasms associated with this mutation have a much higher risk of malignant behavior, with more than 50% of patients developing metastatic disease. There is also an increased risk of renal cell carcinoma and gastrointestinal stromal tumor (GIST).
(Refer to the Familial Paraganglioma Syndrome section in the PDQ summary on Genetics of Endocrine and Neuroendocrine Neoplasias for more information.) - Other syndromes:
- Carney triad syndrome is a condition that includes three tumors: paraganglioma, GIST, and pulmonary chondromas. Pheochromocytomas and other lesions such as esophageal leiomyomas and adrenocortical adenomas have also been described. The syndrome primarily affects young women, with a mean age of 21 years at time of presentation. Approximately one-half of the patients present with paraganglioma or pheochromocytoma, although multiple lesions occur in approximately only 20% of the cases. About 20% of the patients have all three tumor types; the remainder have two of the three, most commonly GIST and pulmonary chondromas. This triad doesn't appear to run in families; however, approximately 10% of the patients have germline variants in the SDHA, SDHB or SDHC genes.[50,51]
- Carney-Stratakis syndrome (Carney dyad syndrome) is a condition that includes paraganglioma and GIST, but not pulmonary chondromas. It is inherited in an autosomal dominant manner with incomplete penetrance. It is equally common in men and women, with an average age of 23 years at presentation. Most patients with this syndrome have been found to carry germline mutations in the SDHB, SDHC, or SDHD genes.[51]
- Other susceptibility genes recently discovered include KIF1B-beta, EGLN1/PHD2, TMEM127, SDHA, and MAX.[49]
Immunohistochemical SDHB staining may help triage genetic testing; tumors of patients with SDHB, SDHC, and SDHD mutations have absent or very weak staining, while sporadic tumors and those associated with other constitutional syndromes have positive staining.[52,53] Therefore, immunohistochemical SDHB staining can help identify potential carriers of a SDH mutation early, obviating the need for extensive and costly testing of other genes. Younger patients have a higher incidence of bilateral adrenal pheochromocytoma and extra-adrenal paraganglioma, and a germline mutation can be identified in close to 60% of patients.[43] Therefore, genetic counseling and testing is always recommended in young patients. Clinical Presentation Patients with pheochromocytoma and sympathetic extra-adrenal paraganglioma usually present with the following symptoms of excess catecholamine production: - Hypertension.
- Headache.
- Perspiration.
- Palpitations.
- Tremor.
- Facial pallor.
These symptoms are often paroxysmal, although sustained hypertension between paroxysmal episodes occurs in more than one-half of patients. These symptoms can also be induced by exertion, trauma, induction of anesthesia, resection of the tumor, consumption of foods high in tyramine (e.g., red wine, chocolate, cheese), or urination (in cases of primary tumor of the bladder).[44] Parasympathetic extra-adrenal paragangliomas do not secrete catecholamines and usually present as a neck mass with symptoms related to compression, but also may be asymptomatic and diagnosed incidentally.[44] The pediatric and adolescent patient appears to present with symptoms similar to those of the adult patient, although with a more frequent occurrence of sustained hypertension.[54] The clinical behavior of paraganglioma and pheochromocytoma appears to be more aggressive in children and adolescents and metastatic rates of up to 50% have been reported.[43,45,54] Genetics Studies of germline mutations in young patients with pheochromocytoma or paraganglioma have further characterized this group of neoplasms, as follows: - In a study of 49 patients younger than 20 years with a paraganglioma or pheochromocytoma, 39 (79%) had an underlying germline mutation that involved the SDHB (n = 27; 55%), SDHD (n = 4; 8%), VHL (n = 6; 12%), or NF1 (n = 2; 4%) gene.[43] The incidence and type of mutation correlated with the site and extent of disease.
- The germline mutation rates for patients with nonmetastatic disease were lower than those observed in patients who had evidence of metastases (64% vs. 87.5%).
- Among patients with metastatic disease, the incidence of SDHB mutations was very high (72%) and most presented with disease in the retroperitoneum; five died of their disease.
- All patients with SDHD mutations had head and neck primary tumors.
- In another study, the incidence of germline mutations involving RET, VHL, SDHD and SDHB in patients with nonsyndromic paraganglioma was 70% for patients younger than 10 years and 51% among those aged 10 to 20 years.[55] In contrast, only 16% of patients older than 20 years had an identifiable mutation.[55]
It is important to note that these two studies did not include systematic screening for other genes that have been recently described in paraganglioma and pheochromocytoma syndromes, such as KIF1B-beta, EGLN1/PHD2, TMEM127, SDHA, and MAX (refer to Table 9). - A retrospective analysis from the European-American-Pheochromocytoma-Paraganglioma-Registry identified 177 patients with paraganglial tumors who were diagnosed before age 18 years.[56][Level of evidence: 3iiA]
- Eighty percent of registrants had germline mutations (49% with VHL, 15% with SDHB, 10% with SDHD, 4% with NF1, and one patient each with RET, SDHA, and SDHC).
- A second primary paraganglial tumor developed in 38% of patients, with increasing frequency over time, reaching 50% at 30 years from initial presentation.
- Prevalence of second tumors was higher in patients with hereditary disease. Sixteen patients (9%) with hereditary disease had malignant tumors, ten at initial presentation and another six during follow-up. Malignancy was associated with SDHB mutations. Eight patients (5%) died, all of whom had a germline mutation. Mean life expectancy was 62 years for patients with hereditary disease.
These findings suggest that younger patients with extra-adrenal nonsyndromic pheochromocytoma and paraganglioma are at high risk of harboring SDHB mutations and that this phenotype is associated with an earlier age of onset and a high rate of metastatic disease. Early identification of young patients with SDHB mutations using radiographic, serologic, and immunohistochemical markers could potentially decrease mortality and identify other family members who carry a germline SDHB mutation. Diagnostic Evaluation The diagnosis of paraganglioma and pheochromocytoma relies on the biochemical documentation of excess catecholamine secretion coupled with imaging studies for localization and staging: - Biochemical testing: Measurement of plasma-free fractionated metanephrines (metanephrine and normetanephrine) is usually the diagnostic tool of choice when the diagnosis of a secreting paraganglioma or pheochromocytoma is suspected. A 24-hour urine collection for catecholamines (epinephrine, norepinephrine, and dopamine) and fractionated metanephrines can also be performed for confirmation.[57,58]
Catecholamine metabolic and secretory profiles are impacted by hereditary background; both hereditary and sporadic paraganglioma and pheochromocytoma differ markedly in tumor contents of catecholamines and corresponding plasma and urinary hormonal profiles. About 50% of secreting tumors produce and contain a mixture of norepinephrine and epinephrine, while most of the rest produce norepinephrine almost exclusively, with occasional rare tumors producing mainly dopamine. Patients with epinephrine-producing tumors are diagnosed later (median age, 50 years) than those with tumors lacking appreciable epinephrine production (median age, 40 years). Patients with MEN2 and NF1 syndromes, all with epinephrine-producing tumors, are typically diagnosed at a later age (median age, 40 years) than are patients with tumors that lack appreciable epinephrine production secondary to mutations of VHL and SDH (median age, 30 years). These variations in ages at diagnosis associated with different tumor catecholamine phenotypes and locations suggest origins of paraganglioma and pheochromocytoma for different progenitor cells with variable susceptibility to disease-causing mutations.[59,60] - Imaging: Imaging modalities available for the localization of paraganglioma and pheochromocytoma include the following:
- Computed tomography (CT).
- Magnetic resonance imaging.
- Iodine I-123 or iodine I-131-labeled metaiodobenzylguanidine (123/131 I-mIBG) scintigraphy.
- Fluorine F-18 6-fluorodopamine (6-[18 F]FDA) positron emission tomography (PET).
For tumor localization, 6-[18 F]FDA PET and 123/131 I-mIBG scintigraphy perform equally well in patients with nonmetastatic paraganglioma and pheochromocytoma, but metastases are better detected by 6-[18 F]FDA PET than by 123/131 I-mIBG.[61] Other functional imaging alternatives include indium In-111 octreotide scintigraphy and fluorodeoxyglucose F-18 PET, both of which can be coupled with CT imaging for improved anatomic detail.
Treatment Treatment of paraganglioma and pheochromocytoma is surgical. For secreting tumors, alpha- and beta-adrenergic blockade must be optimized before surgery. For patients with metastatic disease, responses have been documented to some chemotherapeutic regimens such as gemcitabine and docetaxel or different combinations of vincristine, cyclophosphamide, doxorubicin, and dacarbazine.[62,63,64] Chemotherapy may help alleviate symptoms and facilitate surgery, although its impact on overall survival (OS) is less clear. Responses have also been obtained to high-dose 131 I-mIBG and sunitinib.[65,66] Skin Cancer (Melanoma, Basal Cell Carcinoma [BCC], and Squamous Cell Carcinoma [SCC]) (Refer to the PDQ summary on Genetics of Skin Cancer for more information about specific gene mutations and related cancer syndromes and the Intraocular (Uveal) Melanoma section of this summary for information about uveal melanoma in children.) Melanoma Incidence Melanoma, although rare, is the most common skin cancer in children, followed by BCCs and SCCs.[67,68,69,70,71,72,73,74] In a retrospective study of 22,524 skin pathology reports in patients younger than 20 years, investigators identified 38 melanomas, 33 of which occurred in patients aged 15 to 19 years. Study investigators reported that the number of lesions that needed to be excised to identify one melanoma was 479.8, which is 20 times higher than in the adult population.[75] It is estimated that approximately 400 cases of melanoma are diagnosed each year in patients younger than 20 years in the United States, accounting for less than 1% of all new cases of melanoma.[76] Melanoma annual incidence in the United States (2002-2006) increases with age, as follows:[77,78] - Children younger than 10 years: 1 to 2 cases per 1 million.
- Children aged 10 to 14 years: 4.1 cases per 1 million.
- Children aged 15 to 19 years: 16.9 cases per 1 million.
Melanoma accounts for about 6% of all cancers in children aged 15 to 19 years.[79] The incidence of pediatric melanoma increased by an average of 2% per year between 1973 and 2009.[78] The increased incidence was especially notable in females between the ages of 15 and 19 years. Increased exposure to ambient ultraviolet (UV) radiation increases the risk of the disease. However, a review of United States Surveillance, Epidemiology, and End Results data from 2000 to 2010 suggested that the incidence of melanoma in children and adolescents decreased over that interval.[80] Risk Factors Conditions associated with an increased risk of developing melanoma in children and adolescents include the following: - Giant melanocytic nevi.[70]
- Xeroderma pigmentosum (a rare recessive disorder characterized by extreme sensitivity to sunlight, keratosis, and various neurologic manifestations).[70]
- Immunodeficiency or immunosuppression.[72]
- Hereditary retinoblastoma.[81]
- Werner syndrome.[82,83]
- Neurocutaneous melanosis. Neurocutaneous melanosis is an unusual condition associated with large or multiple congenital nevi of the skin in association with meningeal melanosis or melanoma; approximately 2.5% of patients with large congenital nevi develop this condition, and those with increased numbers of satellite nevi are at greatest risk.[84,85]
Phenotypic traits that are associated with an increased risk of melanoma in adults have been documented in children and adolescents with melanoma and include the following:[86,87,88,89,90,91,92] - Exposure to UV sunlight.
- Red hair.
- Blue eyes.
- Poor tanning ability.
- Freckling.
- Dysplastic nevi.
- Increased number of melanocytic nevi.
- Family history of melanoma.
Prognosis Pediatric melanoma shares many similarities with adult melanoma, and the prognosis is dependent on stage.[93] As in adults, most pediatric cases (about 75%) are localized and have an excellent outcome.[78,89,94] More than 90% of children and adolescents with melanoma are expected to be alive 5 years after their initial diagnosis.[89,93,95,96] The outcome for patients with nodal disease is intermediate, with about 60% expected to survive long term.[89,94,95] In one study, the outcome for patients with metastatic disease was favorable,[89] but this result was not duplicated in another study from the National Cancer Database.[95] Children younger than 10 years who have melanoma often present with poor prognostic features, are more often non-white, have head and neck primary tumors, thicker primary lesions, a higher incidence of spitzoid morphology vascular invasion and nodal metastases, and more often have syndromes that predispose them to melanoma.[89,93,95,97] The use of sentinel node biopsy for staging pediatric melanoma has become widespread, and the thickness of the primary tumor, as well as ulceration, have been correlated with a higher incidence of nodal involvement.[98] Younger patients appear to have a higher incidence of nodal involvement; this finding does not appear to significantly impact clinical outcome in this population.[97,99] In other series of pediatric melanoma, a higher incidence of nodal involvement did not appear to impact survival.[100,101,102] In a retrospective cohort study from the National Cancer Data Base, all records of patients with an index diagnosis of melanoma from 1998 to 2011 were reviewed. The data were abstracted from medical records, operative reports, and pathology reports and did not undergo central review. A total of 350,928 patients with adequate information were identified; 306 patients were aged 1 to 10 years (pediatric), and 3,659 patients were aged 11 to 20 years (adolescent).[103] Pediatric patients had longer OS than did adolescent patients (hazard ratio [HR], 0.50; 95% confidence interval [CI], 0.25-0.98) and patients older than 20 years (HR, 0.11; 95% CI, 0.06-0.21). Adolescents had longer OS than did adults. No difference in OS was found between pediatric node-positive patients and node-negative patients. In pediatric patients, sentinel lymph node biopsy and completion of lymph node dissection were not associated with increased OS. In adolescents, nodal positivity was a significant negative prognostic indicator (HR, 4.82; 95% CI, 3.38-6.87).[103] The association of thickness with clinical outcome is controversial in pediatric melanoma.[89,94,95,104,105,106,107,108] In addition, it is unclear why some variables that correlate with survival in adults are not replicated in children. One possible explanation for this difference might be the inclusion of patients who have lesions that are not true melanomas in the adult series, considering the problematic histological distinction between true melanoma and melanocytic lesions with unknown malignant potential (MELTUMP); these patients are not included in pediatric trials.[109,110] Diagnostic Evaluation The diagnostic evaluation of melanoma includes the following: - Biopsy or excision. Biopsy or excision is necessary to determine the diagnosis of any skin cancer. Diagnosis is necessary for decisions regarding additional treatment. Although BCCs and SCCs are generally curable with surgery alone, the treatment of melanoma requires greater consideration because of its potential for metastasis. The width of surgical margins in melanoma is dictated by the site, size, and thickness of the lesion and ranges from 0.5 cm for in situ lesions to 2 cm or more for thicker lesions.[70] To achieve negative margins in children, wide excision with skin grafting may become necessary in selected cases.
- Lymph node evaluation. Examination of regional lymph nodes using sentinel lymph node biopsy has become routine in many centers [111,112] and is recommended in patients with lesions measuring more than 1 mm in thickness or in those whose lesions are 1 mm or less in thickness and have unfavorable features such as ulceration, Clark level of invasion IV or V, or mitosis rate of 1 per mm2 or higher.[111,113,114] However, the indications for this procedure in patients with spitzoid melanomas has not been clearly defined. In a systematic review of 541 patients with atypical Spitz tumors, 303 (56%) underwent sentinel lymph node biopsy and 119 (39%) had a positive sentinel node; additional lymph node dissection in 97 of these patients revealed additional positive nodes in 18 patients (19%).[115] Despite the high incidence of nodal metastases, only six patients developed disseminated disease, questioning the prognostic and therapeutic benefit of this procedure in children with these lesions. In the future, molecular markers may help identify which patients might benefit from this procedure.
Lymph node dissection is recommended if sentinel nodes are involved with tumor, and adjuvant therapy with high-dose interferon alfa-2b for a period of 1 year should be considered in these patients.[70,111,116,117,118] Clinically benign melanocytic lesions can sometimes pose a significant diagnostic challenge, especially when they involve regional lymph nodes.[119,120,121]
The diagnosis of pediatric melanoma may be difficult and many of these lesions may be confused with the so-called MELTUMP.[122] These lesions are biologically different from melanoma and benign nevi.[122,123] The terms Spitz nevus and spitzoid melanoma are also commonly used, creating additional confusion. One retrospective study found that children aged 10 years or older were more likely to present with amelanotic lesions, bleeding, uniform color, variable diameter, and elevation (such as a de novo bump).[124][Level of evidence: 3iiA] Molecular Features Melanoma-related conditions with malignant potential that arise in the pediatric population can be classified into three general groups:[125] - Large/giant congenital melanocytic nevus.
- Spitzoid melanocytic tumors ranging from atypical Spitz tumors to spitzoid melanomas.
- Melanoma arising in older adolescents that shares characteristics with adult melanoma (i.e., conventional melanoma).
The genomic characteristics of each tumor are summarized in Table 10. The genomic landscape of conventional melanoma in children is represented by many of the genomic alterations found in adults with melanoma.[125] A report from the Pediatric Cancer Genome Project observed that 15 cases of conventional melanoma had a high burden of somatic single-nucleotide variations, TERT promoter mutations (12 of 13), and activating BRAF V600 mutations (13 of 15), as well as a mutational spectrum signature consistent with ultraviolet light damage. In addition, two-thirds of the cases had MC1R variants associated with an increased susceptibility to melanoma. The genomic landscape of spitzoid melanomas is characterized by kinase gene fusions involving various genes including RET, ROS1, NTRK1, ALK, MET, and BRAF.[126,127,128] These fusion genes have been reported in approximately 50% of cases and occur in a mutually exclusive manner.[125,127]TERT promoter mutations are uncommon in spitzoid melanocytic lesions and were observed in only 4 of 56 patients evaluated in one series. However, each of the four cases with TERT promoter mutations experienced hematogenous metastases and died of their disease. This finding supports the potential of TERT promoter mutations in predicting aggressive clinical behavior in children with spitzoid melanocytic neoplasms, but further study is needed to define the role of wild-type TERT promoter status in predicting clinical behavior in patients with primary site spitzoid tumors. Large congenital melanocytic nevi are reported to have activating NRAS Q61 mutations with no other recurring mutations noted.[129] Somatic mosaicism for NRAS Q61 mutations has also been reported in patients with multiple congenital melanocytic nevi and neuromelanosis.[130] Table 10. Characteristics of Melanocytic Lesions| Tumor | Affected Gene |
|---|
| Melanoma | BRAF,NRAS,KIT, NF1 | | Spitzoid melanoma | Kinase fusions (RET,ROS,MET,ALK,BRAF,NTRK1) | | Spitz nevus | HRAS;BRAFandNRAS(uncommon) | | Acquired nevus | BRAF | | Dysplastic nevus | BRAF,NRAS | | Blue nevus | GNAQ | | Ocular melanoma | GNAQ | | Congenital nevi | NRAS | Treatment Surgery is the treatment of choice for patients with localized melanoma. Current guidelines recommend margins of resection as follows: - 0.5 cm for melanoma in situ.
- 1.0 cm for melanoma thickness less than 1 mm.
- 1 cm to 2 cm for melanoma thickness of 1.01 mm to 2 mm.
- 2 cm for tumor thickness greater than 2 mm.
Sentinel node biopsy should be considered in patients with thin lesions (≤1 mm) and ulceration, mitotic rate greater than 1 mm2, young age, and in patients with lesions greater than 1 mm with or without adverse features. Young patients have a higher incidence of sentinel node positivity and this feature adversely affects clinical outcomes.[98,102] If the sentinel node is positive, the option to undergo a complete lymph node dissection should be considered. Patients with high-risk primary cutaneous melanoma, such as those with regional lymph node involvement, can be offered the option to receive adjuvant interferon alfa-2b, a therapy that is well tolerated in children.[116,117,131] Trials of other adjuvant therapies, such as BRAF and MEK inhibitors and checkpoint inhibitors, are currently not available for pediatric patients. For patients with metastatic, recurrent, or progressive disease, prognosis is poor. Various agents such as interferon, dacarbazine, temozolomide, sorafenib, or interleukin-2, and biochemotherapy can be used.[132,133,134] The results of pediatric trials that incorporate newer therapies such as vemurafenib and checkpoint inhibitors including ipilimumab and PD-1 inhibitors are not yet available.[135,136] (Refer to the PDQ summary on adult Melanoma Treatment for more information.) Treatment Options Under Clinical Evaluation The following are examples of national and/or institutional clinical trials that are currently being conducted. Information about ongoing clinical trials is available from the NCI website. - NCT02332668 (A Study of Pembrolizumab [MK-3475] in Pediatric Participants With Advanced Melanoma or Advanced, Relapsed, or Refractory PD-L1-Positive Solid Tumors or Lymphoma [MK-3475-051/KEYNOTE-051]): This is a two-part study of pembrolizumab in pediatric participants who have either advanced melanoma or a programmed cell death ligand 1 (PDL1)-positive advanced, relapsed, or refractory solid tumor or lymphoma. Part 1 will find the maximum tolerated dose/maximum administered dose, confirm the dose, and find the recommended phase II dose for pembrolizumab therapy. Part 2 will further evaluate the safety and efficacy at the pediatric recommended phase II dose.
- NCT02304458 (Nivolumab With or Without Ipilimumab in Treating Younger Patients With Recurrent or Refractory Solid Tumors or Sarcomas): This trial is evaluating the side effects and best dose of nivolumab when given with or without ipilimumab to see how well they work in treating younger patients with solid tumors.
- NCT01677741 (A Study to Determine Safety, Tolerability, and Pharmacokinetics of Oral Dabrafenib In Children and Adolescent Subjects): This is a two-part study to determine the safety, tolerability, and pharmacokinetics of oral dabrafenib in children and adolescent patients with advanced BRAF V600 mutation-positive solid tumors. Part 1 will identify the recommended dose and regimen using a dose-escalation procedure. Part 2 will treat four disease-specific cohorts of patients with tumors known to have BRAF V600 activation (pediatric low-grade gliomas, pediatric high-grade gliomas, Langerhans cell histiocytosis, and other tumors such as melanoma and papillary thyroid carcinoma) using the dose and regimen determined in part 1.
BCC and SCC Clinical Presentation Nonmelanoma skin cancers are very rare in children and adolescents. In one series of 28 patients, approximately one-half of patients had predisposing conditions such as Gorlin syndrome, and one-half of patients were exposed to iatrogenic conditions such as prolonged immunosuppression or radiation.[137] BCCs generally appear as raised lumps or ulcerated lesions, usually in areas with previous sun exposure.[138] These tumors may be multiple and exacerbated by radiation therapy.[139] Nevoid BCC syndrome (Gorlin syndrome) is a rare disorder with a predisposition to the development of early-onset neoplasms, including BCC, ovarian fibroma, and desmoplastic medulloblastoma.[140,141,142,143] SCCs are usually reddened lesions with varying degrees of scaling or crusting, and they have an appearance similar to eczema, infections, trauma, or psoriasis. Diagnostic Evaluation Biopsy or excision is necessary to determine the diagnosis of any skin cancer. Diagnosis is necessary for decisions regarding additional treatment. BCCs and SCCs are generally curable with surgery alone and further diagnostic workup is not indicated. Treatment Treatment for nonmelanoma skin cancer is predominantly surgical, either surgical excision or Mohs micrographic surgery.[137] Most BCCs have activation of the hedgehog pathway, generally resulting from mutations in PTCH1.[144] Vismodegib (GDC-0449), a hedgehog pathway inhibitor, has been approved for the treatment of adult patients with BCC.[145,146] It was approved by the U.S. Food and Drug Administration for the treatment of adults with metastatic BCC or with locally advanced BCC that has recurred after surgery or who are not candidates for surgery, and who are not candidates for radiation. This drug also reduces the tumor burden in patients with basal cell nevus syndrome.[147] (Refer to the PDQ summary on adult Skin Cancer Treatment for more information.) Intraocular (Uveal) Melanoma Incidence and Risk Factors Uveal melanoma (iris, ciliary body, choroid) is the most common primary intraocular malignancy (about 2,000 cases diagnosed each year in the United States) and accounts for 5% of all cases of melanoma.[148] This tumor is most commonly diagnosed in older patients, and the incidence peaks at age 70 years.[149] Pediatric uveal melanoma is extremely rare and accounts for 0.8% to 1.1% of all cases of uveal melanoma.[150] A retrospective, multicenter, observational study conducted by the European Ophthalmic Oncology Group from 1968 to 2014 identified 114 children (aged 1-17 years) and 185 young adults (aged 18-25 years) with ocular melanoma at 24 centers.[150] The median age at the time of diagnosis for children was 15.1 years. The incidence of disease increased by 0.8% per year between the ages of 5 and 10 years and 8.8% per year between the ages of 17 and 24 years. Other series have also documented the higher incidence of the disease in adolescents.[151,152] Risk factors include the following:[153,154,155] - Light eye color.
- Fair skin color.
- Inability to tan.
- Oculodermal melanocytosis.
- Presence of cutaneous nevi.
In a European Oncology Group study, 57% of children were females and four had a preexisting condition that included oculodermal melanocytosis (n = 2) and neurofibromatosis (n = 2).[150] In a review of 13 cases of uveal melanoma in the first 2 years of life, four patients had familial atypical melanoma mole syndrome, one patient had dysplastic nevus syndrome, and one patient had café au lait spots.[156] Molecular Features Uveal melanoma is characterized by activating mutations of GNAQ and GNA11, which lead to activation of the mitogen-activated protein kinases pathway (MAPK). In addition, mutations in BAP1 are seen in 84% of metastasizing tumors, whereas mutations in SF3B1 and EIF1AX are associated with a good prognosis.[157,158,159,160,161,162] Treatment and Outcome Treatment for children is similar to the treatment for adults, which includes surgery, radiation therapy, and laser surgery. (Refer to the PDQ summary on Intraocular (Uveal) Melanoma Treatment for information on the treatment of uveal melanoma in adults.) Survival of children appears to be more favorable than that of young adults and adults, suggesting that the biology of ocular melanoma might be different in children.[150,151] Chordoma Incidence Chordoma is a very rare tumor of bone that arises from remnants of the notochord within the clivus, spinal vertebrae, or sacrum. The incidence in the United States is approximately one case per one million people per year, and only 5% of all chordomas occur in patients younger than 20 years.[163] Most pediatric patients have the classical or chondroid variant of chordoma, while the dedifferentiated variant is rare in children.[163,164] Prognosis Younger children appear to have a worse outlook than do older patients.[163,165,166,167,168,169] The survival rate in children and adolescents ranges from about 50% to 80%.[163,166,168] Clinical Presentation Patients usually present with pain, with or without neurologic deficits such as cranial or other nerve impairment. Diagnosis is straightforward when the typical physaliferous (soap-bubble-bearing) cells are present. Differential diagnosis is sometimes difficult and includes dedifferentiated chordoma and chondrosarcoma. Childhood chordoma has been associated with tuberous sclerosis complex.[170] Treatment Standard treatment includes surgery and external radiation therapy, often proton-beam radiation.[168,171] Surgery is not commonly curative in children and adolescents because of difficulty obtaining clear margins and the likelihood of the chordoma arising in the skull base, rather than in the sacrum, making them relatively inaccessible to complete surgical excision. The best results have been obtained using proton-beam therapy (charged-particle radiation therapy) because these tumors are relatively radiation resistant, and radiation-dose conformality with protons allows for higher tumor doses while sparing adjacent critical normal tissues.[172,173]; [168,174][Level of evidence: 3iiA]; [175][Level of evidence: 3iiiDiii] There are only a few anecdotal reports of the use of cytotoxic chemotherapy after surgery alone or surgery plus radiation therapy. Treatment with ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide has been reported with some success.[176,177] The role for chemotherapy in the treatment of this disease is uncertain. Imatinib mesylate has been studied in adults with chordoma on the basis of the overexpression of PDGFR alpha, beta, and KIT in this disease.[178,179] Among 50 adults with chordoma treated with imatinib and evaluable by Response Evaluation Criteria In Solid Tumors (RECIST) guidelines, there was one partial response and 28 additional patients had stable disease at 6 months.[179] The low rate of RECIST responses and the potentially slow natural course of the disease complicate the assessment of the efficacy of imatinib for chordoma.[179] Other tyrosine kinase inhibitors and combinations involving kinase inhibitors have been studied.[180,181,182] Recurrences are usually local but can include distant metastases to lungs or bone. Cancer of Unknown Primary Site Incidence and Clinical Presentation Children represent less than 1% of all solid cancers of unknown primary site and because of the age-related incidence of tumor types, embryonal histologies are more common in this age group.[183] Cancers of unknown primary site present as a metastatic cancer for which a precise primary tumor site cannot be determined.[184] As an example, lymph nodes at the base of the skull may enlarge in relationship to a tumor that may be on the face or the scalp but is not evident by physical examination or by radiographic imaging. Thus, modern imaging techniques may indicate the extent of the disease but not a primary site. Tumors such as adenocarcinomas, melanomas, and embryonal tumors such as rhabdomyosarcomas and neuroblastomas may present in this way. Diagnostic Evaluation For all patients who present with tumors from an unknown primary site, treatment is directed toward the specific histopathology of the tumor and is age-appropriate for the general type of cancer initiated, irrespective of the site or sites of involvement.[184] Studies in adults suggest that PET imaging can be helpful in identifying cancers of unknown primary site, particularly in patients whose tumors arise in the head and neck area.[185] A report in adults using 2-deoxy-2-[18F]-fluoro-D-glucose (18 F-FDG) PET-CT identified 42.5% of primary tumors in a group of cancers of unknown primary site.[186] The use of gene expression profiling and next-generation sequencing can enhance our ability to identify the putative tissue of origin and guide in the selection of targeted agents for specific mutations.[187,188,189,190,191] No pediatric studies have been conducted to date. Treatment Chemotherapy, targeted therapy, and radiation therapy treatments appropriate and relevant for the general category of carcinoma or sarcoma (depending on the histologic findings, symptoms, and extent of tumor) is initiated as early as possible.[192] (Refer to the PDQ summary on adult Carcinoma of Unknown Primary Treatment for more information.) References:
-
de Krijger RR: Endocrine tumor syndromes in infancy and childhood. Endocr Pathol 15 (3): 223-6, 2004.
-
Thakker RV: Multiple endocrine neoplasia--syndromes of the twentieth century. J Clin Endocrinol Metab 83 (8): 2617-20, 1998.
-
Goudet P, Dalac A, Le Bras M, et al.: MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d'étude des Tumeurs Endocrines. J Clin Endocrinol Metab 100 (4): 1568-77, 2015.
-
Romero Arenas MA, Morris LF, Rich TA, et al.: Preoperative multiple endocrine neoplasia type 1 diagnosis improves the surgical outcomes of pediatric patients with primary hyperparathyroidism. J Pediatr Surg 49 (4): 546-50, 2014.
-
Farnebo F, Teh BT, Kytölä S, et al.: Alterations of the MEN1 gene in sporadic parathyroid tumors. J Clin Endocrinol Metab 83 (8): 2627-30, 1998.
-
Field M, Shanley S, Kirk J: Inherited cancer susceptibility syndromes in paediatric practice. J Paediatr Child Health 43 (4): 219-29, 2007.
-
Thakker RV: Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab 24 (3): 355-70, 2010.
-
Sanso GE, Domene HM, Garcia R, et al.: Very early detection of RET proto-oncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children: presence of C-cell malignant disease in asymptomatic carriers. Cancer 94 (2): 323-30, 2002.
-
Alsanea O, Clark OH: Familial thyroid cancer. Curr Opin Oncol 13 (1): 44-51, 2001.
-
Fitze G: Management of patients with hereditary medullary thyroid carcinoma. Eur J Pediatr Surg 14 (6): 375-83, 2004.
-
Puñales MK, da Rocha AP, Meotti C, et al.: Clinical and oncological features of children and young adults with multiple endocrine neoplasia type 2A. Thyroid 18 (12): 1261-8, 2008.
-
Skinner MA, DeBenedetti MK, Moley JF, et al.: Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg 31 (1): 177-81; discussion 181-2, 1996.
-
Brauckhoff M, Gimm O, Weiss CL, et al.: Multiple endocrine neoplasia 2B syndrome due to codon 918 mutation: clinical manifestation and course in early and late onset disease. World J Surg 28 (12): 1305-11, 2004.
-
Sakorafas GH, Friess H, Peros G: The genetic basis of hereditary medullary thyroid cancer: clinical implications for the surgeon, with a particular emphasis on the role of prophylactic thyroidectomy. Endocr Relat Cancer 15 (4): 871-84, 2008.
-
Waguespack SG, Rich TA, Perrier ND, et al.: Management of medullary thyroid carcinoma and MEN2 syndromes in childhood. Nat Rev Endocrinol 7 (10): 596-607, 2011.
-
Kloos RT, Eng C, Evans DB, et al.: Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19 (6): 565-612, 2009.
-
Wells SA Jr, Asa SL, Dralle H, et al.: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25 (6): 567-610, 2015.
-
Skinner MA, Moley JA, Dilley WG, et al.: Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 353 (11): 1105-13, 2005.
-
Skinner MA: Management of hereditary thyroid cancer in children. Surg Oncol 12 (2): 101-4, 2003.
-
Learoyd DL, Gosnell J, Elston MS, et al.: Experience of prophylactic thyroidectomy in multiple endocrine neoplasia type 2A kindreds with RET codon 804 mutations. Clin Endocrinol (Oxf) 63 (6): 636-41, 2005.
-
Guillem JG, Wood WC, Moley JF, et al.: ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol 24 (28): 4642-60, 2006.
-
National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Thyroid Carcinoma. Version 1.2011. Rockledge, Pa: National Comprehensive Cancer Network, 2011. Available online with free subscription. Last accessed April 04, 2017.
-
Lallier M, St-Vil D, Giroux M, et al.: Prophylactic thyroidectomy for medullary thyroid carcinoma in gene carriers of MEN2 syndrome. J Pediatr Surg 33 (6): 846-8, 1998.
-
Dralle H, Gimm O, Simon D, et al.: Prophylactic thyroidectomy in 75 children and adolescents with hereditary medullary thyroid carcinoma: German and Austrian experience. World J Surg 22 (7): 744-50; discussion 750-1, 1998.
-
Skinner MA, Wells SA Jr: Medullary carcinoma of the thyroid gland and the MEN 2 syndromes. Semin Pediatr Surg 6 (3): 134-40, 1997.
-
Heizmann O, Haecker FM, Zumsteg U, et al.: Presymptomatic thyroidectomy in multiple endocrine neoplasia 2a. Eur J Surg Oncol 32 (1): 98-102, 2006.
-
Frank-Raue K, Buhr H, Dralle H, et al.: Long-term outcome in 46 gene carriers of hereditary medullary thyroid carcinoma after prophylactic thyroidectomy: impact of individual RET genotype. Eur J Endocrinol 155 (2): 229-36, 2006.
-
Piolat C, Dyon JF, Sturm N, et al.: Very early prophylactic thyroid surgery for infants with a mutation of the RET proto-oncogene at codon 634: evaluation of the implementation of international guidelines for MEN type 2 in a single centre. Clin Endocrinol (Oxf) 65 (1): 118-24, 2006.
-
Leboulleux S, Travagli JP, Caillou B, et al.: Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: influence of the stage on the clinical course. Cancer 94 (1): 44-50, 2002.
-
Zenaty D, Aigrain Y, Peuchmaur M, et al.: Medullary thyroid carcinoma identified within the first year of life in children with hereditary multiple endocrine neoplasia type 2A (codon 634) and 2B. Eur J Endocrinol 160 (5): 807-13, 2009.
-
Brauckhoff M, Machens A, Lorenz K, et al.: Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: a changing perspective. Ann Surg 259 (4): 800-6, 2014.
-
Decker RA, Peacock ML, Watson P: Hirschsprung disease in MEN 2A: increased spectrum of RET exon 10 genotypes and strong genotype-phenotype correlation. Hum Mol Genet 7 (1): 129-34, 1998.
-
Eng C, Clayton D, Schuffenecker I, et al.: The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276 (19): 1575-9, 1996.
-
Fialkowski EA, DeBenedetti MK, Moley JF, et al.: RET proto-oncogene testing in infants presenting with Hirschsprung disease identifies 2 new multiple endocrine neoplasia 2A kindreds. J Pediatr Surg 43 (1): 188-90, 2008.
-
Skába R, Dvoráková S, Václavíková E, et al.: The risk of medullary thyroid carcinoma in patients with Hirschsprung's disease. Pediatr Surg Int 22 (12): 991-5, 2006.
-
Moore SW, Zaahl MG: Multiple endocrine neoplasia syndromes, children, Hirschsprung's disease and RET. Pediatr Surg Int 24 (5): 521-30, 2008.
-
Wells SA Jr, Robinson BG, Gagel RF, et al.: Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol 30 (2): 134-41, 2012.
-
Fox E, Widemann BC, Chuk MK, et al.: Vandetanib in children and adolescents with multiple endocrine neoplasia type 2B associated medullary thyroid carcinoma. Clin Cancer Res 19 (15): 4239-48, 2013.
-
Wilkes D, Charitakis K, Basson CT: Inherited disposition to cardiac myxoma development. Nat Rev Cancer 6 (2): 157-65, 2006.
-
Carney JA, Young WF: Primary pigmented nodular adrenocortical disease and its associated conditions. Endocrinologist 2: 6-21, 1992.
-
Ryan MW, Cunningham S, Xiao SY: Maxillary sinus melanoma as the presenting feature of Carney complex. Int J Pediatr Otorhinolaryngol 72 (3): 405-8, 2008.
-
Barontini M, Levin G, Sanso G: Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci 1073: 30-7, 2006.
-
King KS, Prodanov T, Kantorovich V, et al.: Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 29 (31): 4137-42, 2011.
-
Lenders JW, Eisenhofer G, Mannelli M, et al.: Phaeochromocytoma. Lancet 366 (9486): 665-75, 2005 Aug 20-26.
-
Waguespack SG, Rich T, Grubbs E, et al.: A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 95 (5): 2023-37, 2010.
-
Welander J, Söderkvist P, Gimm O: Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 18 (6): R253-76, 2011.
-
Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, et al.: Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 16 (2): 391-400, 2009.
-
Ricketts CJ, Forman JR, Rattenberry E, et al.: Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 31 (1): 41-51, 2010.
-
Burnichon N, Cascón A, Schiavi F, et al.: MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 18 (10): 2828-37, 2012.
-
Boikos SA, Xekouki P, Fumagalli E, et al.: Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet 24 (4): 569-73, 2016.
-
Stratakis CA, Carney JA: The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med 266 (1): 43-52, 2009.
-
Gill AJ, Benn DE, Chou A, et al.: Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 41 (6): 805-14, 2010.
-
van Nederveen FH, Gaal J, Favier J, et al.: An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol 10 (8): 764-71, 2009.
-
Pham TH, Moir C, Thompson GB, et al.: Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics 118 (3): 1109-17, 2006.
-
Neumann HP, Bausch B, McWhinney SR, et al.: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346 (19): 1459-66, 2002.
-
Bausch B, Wellner U, Bausch D, et al.: Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer 21 (1): 17-25, 2014.
-
Lenders JW, Pacak K, Walther MM, et al.: Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287 (11): 1427-34, 2002.
-
Sarathi V, Pandit R, Patil VK, et al.: Performance of plasma fractionated free metanephrines by enzyme immunoassay in the diagnosis of pheochromocytoma and paraganglioma in children. Endocr Pract 18 (5): 694-9, 2012 Sep-Oct.
-
Eisenhofer G, Pacak K, Huynh TT, et al.: Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer 18 (1): 97-111, 2011.
-
Eisenhofer G, Timmers HJ, Lenders JW, et al.: Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab 96 (2): 375-84, 2011.
-
Timmers HJ, Chen CC, Carrasquillo JA, et al.: Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 94 (12): 4757-67, 2009.
-
Mora J, Cruz O, Parareda A, et al.: Treatment of disseminated paraganglioma with gemcitabine and docetaxel. Pediatr Blood Cancer 53 (4): 663-5, 2009.
-
Huang H, Abraham J, Hung E, et al.: Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 113 (8): 2020-8, 2008.
-
Patel SR, Winchester DJ, Benjamin RS: A 15-year experience with chemotherapy of patients with paraganglioma. Cancer 76 (8): 1476-80, 1995.
-
Gonias S, Goldsby R, Matthay KK, et al.: Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol 27 (25): 4162-8, 2009.
-
Joshua AM, Ezzat S, Asa SL, et al.: Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab 94 (1): 5-9, 2009.
-
Sasson M, Mallory SB: Malignant primary skin tumors in children. Curr Opin Pediatr 8 (4): 372-7, 1996.
-
Fishman C, Mihm MC Jr, Sober AJ: Diagnosis and management of nevi and cutaneous melanoma in infants and children. Clin Dermatol 20 (1): 44-50, 2002 Jan-Feb.
-
Hamre MR, Chuba P, Bakhshi S, et al.: Cutaneous melanoma in childhood and adolescence. Pediatr Hematol Oncol 19 (5): 309-17, 2002 Jul-Aug.
-
Ceballos PI, Ruiz-Maldonado R, Mihm MC Jr: Melanoma in children. N Engl J Med 332 (10): 656-62, 1995.
-
Schmid-Wendtner MH, Berking C, Baumert J, et al.: Cutaneous melanoma in childhood and adolescence: an analysis of 36 patients. J Am Acad Dermatol 46 (6): 874-9, 2002.
-
Pappo AS: Melanoma in children and adolescents. Eur J Cancer 39 (18): 2651-61, 2003.
-
Huynh PM, Grant-Kels JM, Grin CM: Childhood melanoma: update and treatment. Int J Dermatol 44 (9): 715-23, 2005.
-
Christenson LJ, Borrowman TA, Vachon CM, et al.: Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA 294 (6): 681-90, 2005.
-
Moscarella E, Zalaudek I, Cerroni L, et al.: Excised melanocytic lesions in children and adolescents - a 10-year survey. Br J Dermatol 167 (2): 368-73, 2012.
-
National Cancer Institute: SEER Stat Fact Sheets: Melanoma of the Skin. Bethesda, Md: National Cancer Institute. Available online. Last accessed March 8, 2017.
-
Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. Bethesda, Md: National Cancer Institute, 2009. Also available online. Last accessed April 04, 2017.
-
Wong JR, Harris JK, Rodriguez-Galindo C, et al.: Incidence of childhood and adolescent melanoma in the United States: 1973-2009. Pediatrics 131 (5): 846-54, 2013.
-
Bleyer A, O'Leary M, Barr R, et al., eds.: Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975-2000. Bethesda, Md: National Cancer Institute, 2006. NIH Pub. No. 06-5767. Also available online. Last accessed January 27, 2017.
-
Campbell LB, Kreicher KL, Gittleman HR, et al.: Melanoma Incidence in Children and Adolescents: Decreasing Trends in the United States. J Pediatr 166 (6): 1505-13, 2015.
-
Kleinerman RA, Tucker MA, Tarone RE, et al.: Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol 23 (10): 2272-9, 2005.
-
Shibuya H, Kato A, Kai N, et al.: A case of Werner syndrome with three primary lesions of malignant melanoma. J Dermatol 32 (9): 737-44, 2005.
-
Kleinerman RA, Yu CL, Little MP, et al.: Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol 30 (9): 950-7, 2012.
-
Hale EK, Stein J, Ben-Porat L, et al.: Association of melanoma and neurocutaneous melanocytosis with large congenital melanocytic naevi--results from the NYU-LCMN registry. Br J Dermatol 152 (3): 512-7, 2005.
-
Makkar HS, Frieden IJ: Neurocutaneous melanosis. Semin Cutan Med Surg 23 (2): 138-44, 2004.
-
Heffernan AE, O'Sullivan A: Pediatric sun exposure. Nurse Pract 23 (7): 67-8, 71-8, 83-6, 1998.
-
Berg P, Lindelöf B: Differences in malignant melanoma between children and adolescents. A 35-year epidemiological study. Arch Dermatol 133 (3): 295-7, 1997.
-
Elwood JM, Jopson J: Melanoma and sun exposure: an overview of published studies. Int J Cancer 73 (2): 198-203, 1997.
-
Strouse JJ, Fears TR, Tucker MA, et al.: Pediatric melanoma: risk factor and survival analysis of the surveillance, epidemiology and end results database. J Clin Oncol 23 (21): 4735-41, 2005.
-
Whiteman DC, Valery P, McWhirter W, et al.: Risk factors for childhood melanoma in Queensland, Australia. Int J Cancer 70 (1): 26-31, 1997.
-
Tucker MA, Fraser MC, Goldstein AM, et al.: A natural history of melanomas and dysplastic nevi: an atlas of lesions in melanoma-prone families. Cancer 94 (12): 3192-209, 2002.
-
Ducharme EE, Silverberg NB: Pediatric malignant melanoma: an update on epidemiology, detection, and prevention. Cutis 84 (4): 192-8, 2009.
-
Paradela S, Fonseca E, Pita-Fernández S, et al.: Prognostic factors for melanoma in children and adolescents: a clinicopathologic, single-center study of 137 Patients. Cancer 116 (18): 4334-44, 2010.
-
Brecht IB, Garbe C, Gefeller O, et al.: 443 paediatric cases of malignant melanoma registered with the German Central Malignant Melanoma Registry between 1983 and 2011. Eur J Cancer 51 (7): 861-8, 2015.
-
Lange JR, Palis BE, Chang DC, et al.: Melanoma in children and teenagers: an analysis of patients from the National Cancer Data Base. J Clin Oncol 25 (11): 1363-8, 2007.
-
Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. Bethesda, Md: National Cancer Institute, 2013. Also available online. Last accessed January 27, 2017.
-
Moore-Olufemi S, Herzog C, Warneke C, et al.: Outcomes in pediatric melanoma: comparing prepubertal to adolescent pediatric patients. Ann Surg 253 (6): 1211-5, 2011.
-
Mu E, Lange JR, Strouse JJ: Comparison of the use and results of sentinel lymph node biopsy in children and young adults with melanoma. Cancer 118 (10): 2700-7, 2012.
-
Balch CM, Soong SJ, Gershenwald JE, et al.: Age as a prognostic factor in patients with localized melanoma and regional metastases. Ann Surg Oncol 20 (12): 3961-8, 2013.
-
Gibbs P, Moore A, Robinson W, et al.: Pediatric melanoma: are recent advances in the management of adult melanoma relevant to the pediatric population. J Pediatr Hematol Oncol 22 (5): 428-32, 2000 Sep-Oct.
-
Livestro DP, Kaine EM, Michaelson JS, et al.: Melanoma in the young: differences and similarities with adult melanoma: a case-matched controlled analysis. Cancer 110 (3): 614-24, 2007.
-
Han D, Zager JS, Han G, et al.: The unique clinical characteristics of melanoma diagnosed in children. Ann Surg Oncol 19 (12): 3888-95, 2012.
-
Lorimer PD, White RL, Walsh K, et al.: Pediatric and Adolescent Melanoma: A National Cancer Data Base Update. Ann Surg Oncol 23 (12): 4058-4066, 2016.
-
Rao BN, Hayes FA, Pratt CB, et al.: Malignant melanoma in children: its management and prognosis. J Pediatr Surg 25 (2): 198-203, 1990.
-
Aldrink JH, Selim MA, Diesen DL, et al.: Pediatric melanoma: a single-institution experience of 150 patients. J Pediatr Surg 44 (8): 1514-21, 2009.
-
Tcheung WJ, Marcello JE, Puri PK, et al.: Evaluation of 39 cases of pediatric cutaneous head and neck melanoma. J Am Acad Dermatol 65 (2): e37-42, 2011.
-
Ferrari A, Bisogno G, Cecchetto G, et al.: Cutaneous melanoma in children and adolescents: the Italian rare tumors in pediatric age project experience. J Pediatr 164 (2): 376-82.e1-2, 2014.
-
Stanelle EJ, Busam KJ, Rich BS, et al.: Early-stage non-Spitzoid cutaneous melanoma in patients younger than 22 years of age at diagnosis: long-term follow-up and survival analysis. J Pediatr Surg 50 (6): 1019-23, 2015.
-
Lohmann CM, Coit DG, Brady MS, et al.: Sentinel lymph node biopsy in patients with diagnostically controversial spitzoid melanocytic tumors. Am J Surg Pathol 26 (1): 47-55, 2002.
-
Su LD, Fullen DR, Sondak VK, et al.: Sentinel lymph node biopsy for patients with problematic spitzoid melanocytic lesions: a report on 18 patients. Cancer 97 (2): 499-507, 2003.
-
Shah NC, Gerstle JT, Stuart M, et al.: Use of sentinel lymph node biopsy and high-dose interferon in pediatric patients with high-risk melanoma: the Hospital for Sick Children experience. J Pediatr Hematol Oncol 28 (8): 496-500, 2006.
-
Kayton ML, La Quaglia MP: Sentinel node biopsy for melanocytic tumors in children. Semin Diagn Pathol 25 (2): 95-9, 2008.
-
Ariyan CE, Coit DG: Clinical aspects of sentinel lymph node biopsy in melanoma. Semin Diagn Pathol 25 (2): 86-94, 2008.
-
Pacella SJ, Lowe L, Bradford C, et al.: The utility of sentinel lymph node biopsy in head and neck melanoma in the pediatric population. Plast Reconstr Surg 112 (5): 1257-65, 2003.
-
Lallas A, Kyrgidis A, Ferrara G, et al.: Atypical Spitz tumours and sentinel lymph node biopsy: a systematic review. Lancet Oncol 15 (4): e178-83, 2014.
-
Navid F, Furman WL, Fleming M, et al.: The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer 103 (4): 780-7, 2005.
-
Chao MM, Schwartz JL, Wechsler DS, et al.: High-risk surgically resected pediatric melanoma and adjuvant interferon therapy. Pediatr Blood Cancer 44 (5): 441-8, 2005.
-
Kirkwood JM, Strawderman MH, Ernstoff MS, et al.: Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14 (1): 7-17, 1996.
-
Roaten JB, Partrick DA, Bensard D, et al.: Survival in sentinel lymph node-positive pediatric melanoma. J Pediatr Surg 40 (6): 988-92; discussion 992, 2005.
-
Ludgate MW, Fullen DR, Lee J, et al.: The atypical Spitz tumor of uncertain biologic potential: a series of 67 patients from a single institution. Cancer 115 (3): 631-41, 2009.
-
Busam KJ, Murali R, Pulitzer M, et al.: Atypical spitzoid melanocytic tumors with positive sentinel lymph nodes in children and teenagers, and comparison with histologically unambiguous and lethal melanomas. Am J Surg Pathol 33 (9): 1386-95, 2009.
-
Berk DR, LaBuz E, Dadras SS, et al.: Melanoma and melanocytic tumors of uncertain malignant potential in children, adolescents and young adults--the Stanford experience 1995-2008. Pediatr Dermatol 27 (3): 244-54, 2010 May-Jun.
-
Cerroni L, Barnhill R, Elder D, et al.: Melanocytic tumors of uncertain malignant potential: results of a tutorial held at the XXIX Symposium of the International Society of Dermatopathology in Graz, October 2008. Am J Surg Pathol 34 (3): 314-26, 2010.
-
Cordoro KM, Gupta D, Frieden IJ, et al.: Pediatric melanoma: results of a large cohort study and proposal for modified ABCD detection criteria for children. J Am Acad Dermatol 68 (6): 913-25, 2013.
-
Lu C, Zhang J, Nagahawatte P, et al.: The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol 135 (3): 816-23, 2015.
-
Wiesner T, He J, Yelensky R, et al.: Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 5: 3116, 2014.
-
Lee S, Barnhill RL, Dummer R, et al.: TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci Rep 5: 11200, 2015.
-
Yeh I, Botton T, Talevich E, et al.: Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 6: 7174, 2015.
-
Charbel C, Fontaine RH, Malouf GG, et al.: NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol 134 (4): 1067-74, 2014.
-
Kinsler VA, Thomas AC, Ishida M, et al.: Multiple congenital melanocytic nevi and neurocutaneous melanosis are caused by postzygotic mutations in codon 61 of NRAS. J Invest Dermatol 133 (9): 2229-36, 2013.
-
Kirkwood JM, Manola J, Ibrahim J, et al.: A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res 10 (5): 1670-7, 2004.
-
Gogas HJ, Kirkwood JM, Sondak VK: Chemotherapy for metastatic melanoma: time for a change? Cancer 109 (3): 455-64, 2007.
-
Eton O, Legha SS, Bedikian AY, et al.: Sequential biochemotherapy versus chemotherapy for metastatic melanoma: results from a phase III randomized trial. J Clin Oncol 20 (8): 2045-52, 2002.
-
Middleton MR, Grob JJ, Aaronson N, et al.: Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18 (1): 158-66, 2000.
-
Chapman PB, Hauschild A, Robert C, et al.: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364 (26): 2507-16, 2011.
-
Hodi FS, O'Day SJ, McDermott DF, et al.: Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8): 711-23, 2010.
-
Khosravi H, Schmidt B, Huang JT: Characteristics and outcomes of nonmelanoma skin cancer (NMSC) in children and young adults. J Am Acad Dermatol 73 (5): 785-90, 2015.
-
Efron PA, Chen MK, Glavin FL, et al.: Pediatric basal cell carcinoma: case reports and literature review. J Pediatr Surg 43 (12): 2277-80, 2008.
-
Griffin JR, Cohen PR, Tschen JA, et al.: Basal cell carcinoma in childhood: case report and literature review. J Am Acad Dermatol 57 (5 Suppl): S97-102, 2007.
-
Gorlin RJ: Nevoid basal cell carcinoma syndrome. Dermatol Clin 13 (1): 113-25, 1995.
-
Kimonis VE, Goldstein AM, Pastakia B, et al.: Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 69 (3): 299-308, 1997.
-
Amlashi SF, Riffaud L, Brassier G, et al.: Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer 98 (3): 618-24, 2003.
-
Veenstra-Knol HE, Scheewe JH, van der Vlist GJ, et al.: Early recognition of basal cell naevus syndrome. Eur J Pediatr 164 (3): 126-30, 2005.
-
Caro I, Low JA: The role of the hedgehog signaling pathway in the development of basal cell carcinoma and opportunities for treatment. Clin Cancer Res 16 (13): 3335-9, 2010.
-
Von Hoff DD, LoRusso PM, Rudin CM, et al.: Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 361 (12): 1164-72, 2009.
-
Sekulic A, Migden MR, Oro AE, et al.: Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med 366 (23): 2171-9, 2012.
-
Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al.: Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med 366 (23): 2180-8, 2012.
-
Field MG, Harbour JW: Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol 25 (3): 234-9, 2014.
-
Singh AD, Bergman L, Seregard S: Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am 18 (1): 75-84, viii, 2005.
-
Al-Jamal RT, Cassoux N, Desjardins L, et al.: The Pediatric Choroidal and Ciliary Body Melanoma Study: A Survey by the European Ophthalmic Oncology Group. Ophthalmology 123 (4): 898-907, 2016.
-
Shields CL, Kaliki S, Arepalli S, et al.: Uveal melanoma in children and teenagers. Saudi J Ophthalmol 27 (3): 197-201, 2013.
-
Pogrzebielski A, Orłowska-Heitzman J, Romanowska-Dixon B: Uveal melanoma in young patients. Graefes Arch Clin Exp Ophthalmol 244 (12): 1646-9, 2006.
-
Weis E, Shah CP, Lajous M, et al.: The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol 124 (1): 54-60, 2006.
-
Weis E, Shah CP, Lajous M, et al.: The association of cutaneous and iris nevi with uveal melanoma: a meta-analysis. Ophthalmology 116 (3): 536-543.e2, 2009.
-
Singh AD, De Potter P, Fijal BA, et al.: Lifetime prevalence of uveal melanoma in white patients with oculo(dermal) melanocytosis. Ophthalmology 105 (1): 195-8, 1998.
-
Yousef YA, Alkilany M: Characterization, treatment, and outcome of uveal melanoma in the first two years of life. Hematol Oncol Stem Cell Ther 8 (1): 1-5, 2015.
-
Van Raamsdonk CD, Griewank KG, Crosby MB, et al.: Mutations in GNA11 in uveal melanoma. N Engl J Med 363 (23): 2191-9, 2010.
-
Harbour JW, Onken MD, Roberson ED, et al.: Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330 (6009): 1410-3, 2010.
-
Gupta MP, Lane AM, DeAngelis MM, et al.: Clinical Characteristics of Uveal Melanoma in Patients With Germline BAP1 Mutations. JAMA Ophthalmol 133 (8): 881-7, 2015.
-
Harbour JW, Roberson ED, Anbunathan H, et al.: Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45 (2): 133-5, 2013.
-
Martin M, Maßhöfer L, Temming P, et al.: Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet 45 (8): 933-6, 2013.
-
Van Raamsdonk CD, Bezrookove V, Green G, et al.: Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457 (7229): 599-602, 2009.
-
Hoch BL, Nielsen GP, Liebsch NJ, et al.: Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol 30 (7): 811-8, 2006.
-
McMaster ML, Goldstein AM, Bromley CM, et al.: Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control 12 (1): 1-11, 2001.
-
Coffin CM, Swanson PE, Wick MR, et al.: Chordoma in childhood and adolescence. A clinicopathologic analysis of 12 cases. Arch Pathol Lab Med 117 (9): 927-33, 1993.
-
Borba LA, Al-Mefty O, Mrak RE, et al.: Cranial chordomas in children and adolescents. J Neurosurg 84 (4): 584-91, 1996.
-
Jian BJ, Bloch OG, Yang I, et al.: A comprehensive analysis of intracranial chordoma and survival: a systematic review. Br J Neurosurg 25 (4): 446-53, 2011.
-
Yasuda M, Bresson D, Chibbaro S, et al.: Chordomas of the skull base and cervical spine: clinical outcomes associated with a multimodal surgical resection combined with proton-beam radiation in 40 patients. Neurosurg Rev 35 (2): 171-82; discussion 182-3, 2012.
-
Chambers KJ, Lin DT, Meier J, et al.: Incidence and survival patterns of cranial chordoma in the United States. Laryngoscope 124 (5): 1097-102, 2014.
-
McMaster ML, Goldstein AM, Parry DM: Clinical features distinguish childhood chordoma associated with tuberous sclerosis complex (TSC) from chordoma in the general paediatric population. J Med Genet 48 (7): 444-9, 2011.
-
DeLaney TF, Liebsch NJ, Pedlow FX, et al.: Long-term results of Phase II study of high dose photon/proton radiotherapy in the management of spine chordomas, chondrosarcomas, and other sarcomas. J Surg Oncol 110 (2): 115-22, 2014.
-
Hug EB, Sweeney RA, Nurre PM, et al.: Proton radiotherapy in management of pediatric base of skull tumors. Int J Radiat Oncol Biol Phys 52 (4): 1017-24, 2002.
-
Noël G, Habrand JL, Jauffret E, et al.: Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther Onkol 179 (4): 241-8, 2003.
-
Rombi B, Ares C, Hug EB, et al.: Spot-scanning proton radiation therapy for pediatric chordoma and chondrosarcoma: clinical outcome of 26 patients treated at paul scherrer institute. Int J Radiat Oncol Biol Phys 86 (3): 578-84, 2013.
-
Rutz HP, Weber DC, Goitein G, et al.: Postoperative spot-scanning proton radiation therapy for chordoma and chondrosarcoma in children and adolescents: initial experience at paul scherrer institute. Int J Radiat Oncol Biol Phys 71 (1): 220-5, 2008.
-
Dhall G, Traverso M, Finlay JL, et al.: The role of chemotherapy in pediatric clival chordomas. J Neurooncol 103 (3): 657-62, 2011.
-
Al-Rahawan MM, Siebert JD, Mitchell CS, et al.: Durable complete response to chemotherapy in an infant with a clival chordoma. Pediatr Blood Cancer 59 (2): 323-5, 2012.
-
Casali PG, Messina A, Stacchiotti S, et al.: Imatinib mesylate in chordoma. Cancer 101 (9): 2086-97, 2004.
-
Stacchiotti S, Longhi A, Ferraresi V, et al.: Phase II study of imatinib in advanced chordoma. J Clin Oncol 30 (9): 914-20, 2012.
-
Lindén O, Stenberg L, Kjellén E: Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol 48 (1): 158-9, 2009.
-
Singhal N, Kotasek D, Parnis FX: Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs 20 (10): 953-5, 2009.
-
Stacchiotti S, Marrari A, Tamborini E, et al.: Response to imatinib plus sirolimus in advanced chordoma. Ann Oncol 20 (11): 1886-94, 2009.
-
Pavlidis N, Pentheroudakis G: Cancer of unknown primary site. Lancet 379 (9824): 1428-35, 2012.
-
Kuttesch JF Jr, Parham DM, Kaste SC, et al.: Embryonal malignancies of unknown primary origin in children. Cancer 75 (1): 115-21, 1995.
-
Bohuslavizki KH, Klutmann S, Kröger S, et al.: FDG PET detection of unknown primary tumors. J Nucl Med 41 (5): 816-22, 2000.
-
Han A, Xue J, Hu M, et al.: Clinical value of 18F-FDG PET-CT in detecting primary tumor for patients with carcinoma of unknown primary. Cancer Epidemiol 36 (5): 470-5, 2012.
-
Tothill RW, Li J, Mileshkin L, et al.: Massively-parallel sequencing assists the diagnosis and guided treatment of cancers of unknown primary. J Pathol 231 (4): 413-23, 2013.
-
Varadhachary GR, Talantov D, Raber MN, et al.: Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 26 (27): 4442-8, 2008.
-
Fizazi K, Greco FA, Pavlidis N, et al.: Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 26 (Suppl 5): v133-8, 2015.
-
Greco FA, Lennington WJ, Spigel DR, et al.: Poorly differentiated neoplasms of unknown primary site: diagnostic usefulness of a molecular cancer classifier assay. Mol Diagn Ther 19 (2): 91-7, 2015.
-
Gatalica Z, Millis SZ, Vranic S, et al.: Comprehensive tumor profiling identifies numerous biomarkers of drug response in cancers of unknown primary site: analysis of 1806 cases. Oncotarget 5 (23): 12440-7, 2014.
-
Morris GJ, Greco FA, Hainsworth JD, et al.: Cancer of unknown primary site. Semin Oncol 37 (2): 71-9, 2010.
Changes to This Summary (04 / 04 / 2017)The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above. Head and Neck Cancers Added text to state that a retrospective review of the Surveillance, Epidemiology, and End Results database from 1973 to 2011 identified 2,504 cases of papillary thyroid carcinoma in patients aged 20 years and younger. The incidence of papillary thyroid carcinoma increased over this interval by roughly 2% each year (cited Golpanian et al. as reference 53). Abdominal Cancers Added text to state that despite the increased risk of multiple malignancies in families with Lynch syndrome, the risk of malignant neoplasms during childhood in those families does not seem to be increased when compared with the risk in children from non-Lynch syndrome colorectal carcinoma families (cited Heath et al. as reference 107). Added text about the findings of a single-institution retrospective review that identified 45 cases of carcinoid tumors in children and adolescents between 2003 and 2016 (cited Degnan et al. as reference 115 and level of evidence 3iiDii). Added text to state that larger tumor size has been associated with a higher risk of recurrence of nonappendiceal neuroendocrine tumors. This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages. About This PDQ SummaryPurpose of This Summary This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of unusual cancers of childhood. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions. Reviewers and Updates This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH). Board members review recently published articles each month to determine whether an article should: - be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary. The lead reviewers for Unusual Cancers of Childhood Treatment are: - Denise Adams, MD (Children's Hospital Boston)
- Karen J. Marcus, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (AFLAC Cancer Center and Blood Disorders Service of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- R Beverly Raney, MD (Consultant)
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries. Levels of Evidence Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations. Permission to Use This Summary PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]." The preferred citation for this PDQ summary is: PDQ® Pediatric Treatment Editorial Board. PDQ Unusual Cancers of Childhood Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/childhood-cancers/hp/unusual-cancers-childhood-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389315] Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images. Disclaimer Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page. Contact Us More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us. Last Revised: 2017-04-04 Last modified on: 8 September 2017
|
|